Антибіотики (властивості, застосування, взаємодія) - М.П. Черенько 1999
Вади розвитку та виродливості
ВИЗНАЧЕННЯ, Класифікація, Етіологія, Патогенез
Вади розвитку, чи аномалії, — це природжені відхилення за межі нормальних варіантів у анатомічній будові (формі, розмірах, числі) тканин та органів людини, які здебільшого супроводжуються порушеннями їх функцій чи навіть загрожують життєздатності організму. Вищі ступені природжених вад розвитку багатьох органів та систем організму називають виродливістю.
Вивчення походження та патології вад складає окрему дисципліну медичної науки — тератологію (гр. teratos — чудовисько, logos — поняття, вчення).
Вади розвитку — поширений вид патології, питома вага якого в загальній популяції коливається в різних країнах, за даними ВООЗ, від 2,7 до 16,3 % і має тенденцію в останні десятиріччя до росту.
Існує величезна кількість вад, як видимих, так і невидимих, морфологічного (макро- і мікроскопічні) та біохімічного (природжені порушення обміну речовин) характеру. Прикладами останніх є агаммаглобулінемія, галактозурія, гемофілія А, альбінізм, алкаптонурія та багато інших. У основі переважної більшості біохімічних вад лежить порушення в ензимних системах організму. Біохімічні вади в організмі людини вивчають терапевти.
До компетенції хірургів належать морфологічні вади макроскопічного типу, вади ж гістоморфологічного (мікроскопічного) характеру потрапляють в поле зору хірургів лише тоді, коли вони є джерелом розвитку патологічного процесу, насамперед пухлин. До таких мікроскопічних вад належать різноманітні за будовою та локалізацією патологічні клітинні скупчення в тканинах та органах, які мають загальну назву гетеротопії. Серед них найвідоміші так звані гамартії (від гр. chamartano — помиляюсь, та choriso — відділяю), з яких походять пухлини гамартома та хористома, прикладом їх відповідно є капілярна ангіома шкіри та хондрома легень.
Досконалої класифікації природжених вад немає через різноманітний їх характер, локалізацію, походження та тяжкість анатомо-фізіологічних розладів організму. Залежно від класифікаційного критерію виділяють низку груп вад. За локалізацією в організмі вони можуть бути поділені на:
1) зовнішні (розщеплення верхньої Губи, атрезія задньопрохідного отвору тощо);
2) внутрішні (дефект перегородки між шлуночками чи передсердями серця; Пілоростеноз — різке звуження воротарного отвору шлунка та ін.);
3) комбіновані чи змішані (щілина губи і піднебіння та діафрагмальна грижа та ін.).
За кількістю вад у людини їх ділять таким чином :
1) вади поодинокі — органні чи системні (гіпоспадія та ін.; хондродисплазія тощо);
2) вади множинні — органні чи системні (щілина піднебіння та Мозкова грижа; вади в межах травної та серцево-судинної систем; нервової та ендокринної тощо);
3) вади одного організму та вади двох організмів (зрощені близнюки чи подвійні виродливості).
З клінічної точки зору, вади часто класифікуються також відповідно до анатомо-фізіологічного поділу організму на системи, ділянки та органи тіла (вади ЦНС, вади органів травного каналу, вади сечовидільної системи, вади обличчя, вади шкіри тощо).
Розрізняють також вади первинні та вторинні — залежно від послідовності їх появи. Так, первинний дефект розвитку лікворних комунікаційних шляхів головного мозку (їх звуження чи відсутність формування) породжує вторинну патологію — гідроцефалію з деформацією черепа.
У основі походження вад лежать різноманітні порушення процесів та розвитку тканин і органів, а саме:
1) агенезія та аплазія — повна відсутність органа (відповідно з відсутністю і його закладки чи з наявністю лише судинної ніжки);
2) гіпоплазія — недостатній розвиток маси органа, окремих його частин або всього тіла (гіпотрофія);
3) природжена гіпертрофія органа (гігантизм чи слоновість) за рахунок збільшення його об’єму або кількості клітинної маси (гіперплазії);
4) етеротопія — наявність комплексу клітин, частин Тканини або органа в тканинах чи органах (гамартія та хористія);
5) ектопія та дистопія — ненормальна локалізація органа (язикова чи внутрішньогрудинна ектопія щитовидної залози);
6) стеноз — звуження діаметра каналу чи порожнини легеневої Артерії, стравоходу, кишки тощо;
7) атрезія — зарощення отворів чи каналів органів (атрезія прямої кишки чи задньопрохідного отвору, стравоходу);
8) нерозділення органів чи організмів (Синдактилія — зрощення пальців, зрощення між собою близнюків різними частинами тіла, зокрема грудьми — торакопаги чи стернопаги, головами — craniopagus, крижами — pygopagus тощо);
9) персистування — збереження після народження проток, які звичайно функціонують лише в Ембріональний період (відкрита артеріальна протока між аортою та легеневою артерією, збереження відкритої пупково-мезентеріальної чи пупково-сечоміхурової протоки);
10) подвоєння органа чи частини його (подвоєння окремих петель чи відрізків кишечнику, пальців — полідактилія, матки, очеревини тощо);
11) інверсія розташування органа чи органів унаслідок порушення процесу повороту їх — серця, кишки (декстрокардія; situs viscerus inversus — органів черевної порожнини);
12) зупинка зрощення парних сегментів органа чи боків (країв) ембріональних трубок, каналів, порожнини тощо (щелепних відростків, черевної порожнини, сечовивідного каналу); передчасне заростання чи закриття порожнини тіла, отворів органів тощо;
13) атавізм — поява у людини тканинних структур у місцях, де вони є у тварин (наприклад, оволосіння всього обличчя чи окремих ділянок тіла; рудименти хвоста — хвостових хребців; іхтіоз — лускоподібна Шкіра; полімастія та політелія — збільшення числа молочних залоз, сосків; справжній Гермафродитизм тощо).
Залежно від етіології Природжені вади розвитку умовно ділять на три групи: спадкові, екзогенні та мультифакторіальні (Г.І. Лазюк, 1991).
Спадкові вади пов’язані зі структурними дефектами генів та хромосом (мутаціями). Екзогенні вади спричинені дією тератогенних чинників на ембріон та плід. Вади мультифакторіальні зумовлюються поєднаною дією генетичних та екзогенних причин, кожна з яких самостійно нездатна спричинити ваду.
Всі численні етіологічні чинники вад розвитку можна розділити на дві групи — ендогенні та екзогенні.
До групи ендогенних чинників належать: Мутації спадкових структур, ендокринні захворювання, перезрілість статевих клітин та немолодий вік батьків.
Групу екзогенних чинників складають: фізичні, хімічні та біологічні.
Мутації спадкових структур — генів та хромосом — можуть бути як стійкими, так і нестійкими, і відбуваються як спонтанно, так і під впливом дії на спадкові структури різних екзогенних чинників (так званий індукований мутагенез). Хоч Генні мутації бувають значно рідше, ніж хромосомні (мікроскопічні порушення хромосом) та геномні (порушення кількості хромосом), але більшість природжених вад є наслідком саме генних мутацій, зокрема полігенних. Багато авторів вважають мутації головною причиною вад. На їхню думку, екзогенні чинники зумовлюють вади розвитку також через вплив на чинники спадковості, тобто через мутагенні ефекти.
Широке вивчення проблеми походження вад розвитку, проведене в останні роки, дає підставу стверджувати, що переважна більшість вад розвитку має полігенно-мультифакторіальне походження.
Мутації спадкових утворень (генів, хромосом) можуть відбуватись спонтанно, в процесі фізіологічної діяльності організму, як під дією різних ендогенних метаболітів, так і внаслідок шкідливого впливу численних екзогенних чинників.
Ендокринні захворювання, порушення секреції гормонів (гіперпродукція стероїдів наднирковими залозами плода, Цукровий діабет у матері тощо) також призводять до розвитку вад (вірильний синдром, діабетичні ембріопатії тощо). Перезрівання статевих клітин (тривале їх існування до злиття в гамету) та немолодий вік батьків теж нерідко є причинами природжених вад розвитку.
Серед численних вад, зумовлених мутаціями спадкових структур, зокрема хромосом та генома, такі ендокринні вади, як синдром Клейнфелтера (фенотип 47 XXY) та синдром Шерешевського—Тернера (фенотип 45), хвороба Дауна (трисомія 21 Хромосоми), окремі вади серця тощо.
Серед зовнішніх причин, що сприяють розвитку природжених вад, насамперед треба назвати прогресуюче погіршення екології, незадовільний стан довкілля (забруднення повітря, води та землі відходами промислової діяльності людини тощо). При цьому негативні чинники навколишнього середовища можуть діяти як прямо на Організм людини, зокрема на плід, так і опосередковано, трансформуючи його внутрішнє середовище. Ця обставина робить практично неможливим чітке розмежування зовнішніх та внутрішніх тератогенних агентів. Безпосередні причини більшості вад розвитку ще не з’ясовані, і далеко не всі несприятливі чинники навколишнього і внутрішнього середовища вивчені достатньою мірою. Але вплив багатьох із них на розвиток вад добре відомий.
Найбільшу роль у генезі вад розвитку відіграють фізичні чинники, особливо рентгенівське та радіоактивне опромінення, деякі хімічні речовини, медичні препарати, інфекційні захворювання, дефіцит амінокислот, білків, вітамінів та інших поживних речовин у дієті чи організмі вагітної, порушення гормонального балансу в її організмі, Гіпоксія, механічні впливи на плід тощо.
Негативна дія рентгенівського та радіоактивного опромінення на організм живих істот, особливо спадковий механізм (Геном) їх, добре відома від часів відкриття цих променів. Але тератогенна дія їх особливо стала відомою після вивчення наслідків атомного бомбардування Міст Японії Хіросими та Нагасакі.
Подальші експериментальні роботи в цьому напрямку та спостереження над людьми в інших зонах, де населення зазнало радіоактивного опромінення, підтверджують Висновки, зроблені японськими вченими. Треба зазначити, що для радіоактивних променів не існує межового рівня тератогенності, тобто дія їх навіть у найменшій дозі потенційно тератогенна.
З хімічних чинників особливо тератогенним вважається етиловий спирт. Алкоголізм матерів є причиною вад розвитку у 30 % дітей, серед яких вади серця спостерігаються у 30—49 % випадків (Н.П. Білоконь, В.П. Подзолков, 1991). Хронічна інтоксикація організму алкоголем спричинює пошкодження біологічної структури статевих клітин, яке часто призводить до гіпогонадизму, крипторхізму, природжених гриж, вад серця та нервової системи тощо.
Багато інших хімічних сполук (саліцилати, антиметаболіти, цитостатики, інсектициди, оксид анти, сполуки арсену, хрому тощо), серед яких багато і медичних препаратів (наркотики, транквілізатори, гормональні препарати тощо), теж здатні спричинити порушення розвитку плода. Яскравим прикладом такої дії фармацевтичних препаратів є вплив талідоміду, який був синтезований у Німеччині та поширений в 57—60-х роках у Західній Європі як снотворний засіб. У жінок, які користувались цим препаратом, часто народжувалися діти з вадами розвитку кінцівок.
Численні інфекційні, особливо вірусні, захворювання також стають причиною розвитку багатьох вад, зокрема вад серця, черепа та мозку, статевих залоз. Серед них такі інфекційні захворювання, як Грип, кір, корова краснуха, Токсоплазмоз, епідемічний паротит та гепатит, ревмокардит та ін.
Неінфекційні захворювання матері, які супроводжуються розвитком у неї гіпоксемії (Серцева недостатність, анемія), зумовлюючи гіпоксію плода, сприяють розвитку в нього вад.
Парціальні форми голодування, зокрема дефіцит амінокислот і білків, вітамінів (фолієва кислота та ін.), можуть бути причиною розвитку вад, особливо нервової системи.
Багато вад виникає внаслідок дії на плід механічних причин — тиску амніотичних оболонок та перетинок, маловоддя, механічного походження тощо. Серед порушень цього походження — вади кінцівок, нервової системи, Кривошия тощо.
Природжені вади є наслідком порушення розвитку ембріона та плода на різних його етапах — гаметогенезу, Запліднення, ембріонального морфогенезу (виникнення спеціалізованих тканин з малодиференційованих клітин та розвиток органів — Органогенез) та постембріонального періоду.
Головними клітинними механізмами тератогенезу є порушення їх розмноження, Міграції та диференціювання. Наслідками розладу згаданих процесів є такі поширені вади, як агенезія та гіпоплазія органів, порушення зрощування між собою окремих зародкових утворень, що веде до дизрафій (щілин), гетеротопія, функціональна незрілість тканин та органів і персистування ембріональних структур (незарощення проток) тощо.
Слід зазначити, що хоч вади розвитку можуть виникати протягом усього внутрішньоутробного періоду (гамето-, бласто-, ембріо- та фегопатії), але найчастіше вони утворюються в так звані критичні періоди, коли зародок дуже чутливий до шкідливих агентів середовища. Це перші 6 тиж ембріогенезу (вади кінця 2-го тижня цього періоду несумісні з життям; вади, що виникають на 3—6-му тижні, переважно сумісні з життям).
Ці періоди збігаються з найінтенсивнішим періодом формування органів та систем. Що раніше виникає вада, то вона серйозніша. Оскільки травмівний чинник може зумовити ваду розвитку органа лише до закінчення формування його, а терміни формування різних органів різні, важливим для теорії та практики тератології є розроблене Е. Schwalbe (1906) вчення про так звані термінаційні тератогенетичні періоди, тобто хронологічні рамки, в межах яких може виникати вада розвитку органа.
Так, термінаційний період для нерозділених близнюків становить перші 2 тиж після запліднення, а для дефекту міжпередсердної перегородки — до 44-ї доби вагітності. Крипторхізм, незарощення проток — вади пізнішого періоду (фетопатії).
ЗОВНІШНІ ВАДИ
Вадн розвитку ЦНС. Численні за варіантами і ступенем анатомофізіологічних порушень вади ЦНС — головного та спинного мозку — найчастіше виникають у разі затримки закриття ембріональної нервової трубки, яке в нормі відбувається наприкінці 4-го тижня внутрішньоутробного розвитку, або, рідше, за передчасного закриття цієї трубки.
Формування вад спостерігається переважно на кінцях нервової трубки — передньому (головному) чи задньому (каудальному). Передчасне закриття переднього кінця нейропори призводить до зупинки розвитку головного мозку — акранії та аненцефалії (відсутності утворення черепа та мозку).
До тяжких вад раннього періоду ембріогенезу належать також циклопія — наявність на деформованому черепі однієї орбіти зі злитими та недорозвинутими очними яблуками в ній і носом у формі паростка шкіри над оком і синотія з агнатією — зрощення вушних раковин своїми нижніми кінцями в ділянці нижньої щелепи, якої немає.
Затримка в часі закриття переднього кінця нервової трубки супроводжується незарощенням кісток черепа та виходом через кістковий дефект мозкової грижі. Остання може складатися лише з оболонки мозку (м’якої) і мати форму грижового мішка з черепно-мозковою рідиною (ліквором) в ньому — така грижа називається менінгоцеле та гідроменінгоцеле, або включати також і мозкову тканину — тоді вона називається енцефалоцеле. Мозкові грижі бувають переважно задні, потиличні (мал. 116, б), рідше — передні, лобні, які виходять над основою носа (мал. 116, а) чи біля внутрішнього куточка ока.
Вади черепа та головного мозку можуть також зумовлюватися передчасним зрощенням між собою черепних кісток — закриттям одного чи багатьох черепно-мозкових швів (сrаnіоsynostosis). Це веде до деформації черепа (мікроцефалії та оксицефалії) та порушення функції мозку головним чином через гідроцефалію (підвищення тиску ліквору в мозку та черепі).
Мікроцефалія виявляється малими розмірами голови та мозку, атрофією мозку та розтягненням його ліквором.
Ознаки оксицефалії — диспропорційний за формою Череп, переважно баштовий (високий, вузький, витягнутий в сагітальному напрямку), недостатність інтелекту, сліпота, дилатація мозкових шлуночків.
Серед вад черепа та мозку значне місце належить природженій гідроцефалії. Під нею розуміють стан, за якого в період народження або відразу після нього інтракраніально збільшується маса цереброспінальної рідини. Ознаки природженої гідроцефалії: збільшені розміри голови, широко відкриті тім’ячка та шви черепа, різко розширені внутрішньомозкові шлуночки з великою кількістю в них ліквору. Це — гідромакроцефалія. Причинами гідроцефалії можуть бути насамперед обструктивні явища в лікворних комунікаціях, а також підвищена продукція ліквору та дисбаланс між його продукцією і всмоктуванням у вени.
Блокада лікворних шляхів може бути зумовлена недорозвиненістю (атрезією) отворів між шлуночками або епідидимітом інфекційного чи токсичного генезу.
Серед інфекцій, зокрема, велика роль у розвитку вад раніше відводилася токсоплазмозу, зараз це вважається перебільшенням.
Лікування гідроцефалії полягає в ранній операції шунтування лікворної системи черепа в черевну порожнину. У разі раннього виконання операції та ретельного догляду за станом шунта можна досягти добрих наслідків лікування.
У разі затримки закриття каудального кінця ембріональної нервової трубки може виникати розщеплення хребетного стовпа (Rachischisis) на всій його відстані (внаслідок відсутності формування дорсальних дуг хребців), в жолобі якого лежить незакрита нервова трубка, або ж мієлоцеле (myelocele). Вада подібна до попередньої, але з тою різницею, що в разі останньої частина дорсальних дужок сформована (переважно в грудному відділі), а нервова трубка на всій її відстані частково сформована або закрита.
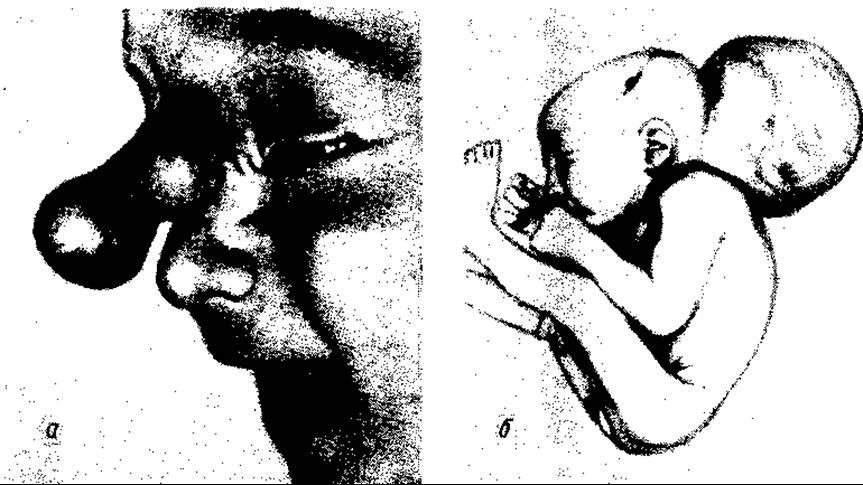
Мал. 116. Мозкова грижа:
а — передня носова (М.Б. Ситковський та співавт.); б — потилична
Крім цих двох тяжких, що несумісні з життям, вад, частіше зустрічаються менінгоцеле (meningocele) — випинання через локальний дефект у хребетному стовпі та твердій мозковій оболонці м’якої Оболонки спинного мозку в формі пухиря, наповненого спинномозковою рідиною, під шкіру — та менінгомієлоцеле (meningomyelocele) — випинання через дефект хребетного стовпа як м’якої оболонки спинного мозку, так і самого мозку під шкіру (подібно до енцефалоцеле).
Менінго- та менінгомієлоцеле спостерігаються переважно в поперековому відділі хребетного стовпа.
Частим варіантом вад спинного мозку та хребетного стовпа є так званий прихований спінальний дизрафізм, чи приховане розщеплення хребетного стовпа (spina bifida occulta), який полягає в дефекті дужок хребетного стовпа в попереково-крижовому відділі його, закритому сполучною тканиною (без випинання оболонки спинного мозку та мозку).
Цю ваду виявляють або випадково, під час обстеження хворого з іншого приводу, або, частіше, в зв’язку з наявністю у хворих на шкірі в зоні дефекту хребетного стовпа обмеженого гіпертрихозу, капілярної гемангіоми, ліпоми, дермальних синусів чи сколіозу.
До цієї групи вад (менінгомієлоцеле) належить також низка інших порушень. У тому числі дистензія центральна, однобічні щілини губи. Щілина піднебіння може бути як у поєднанні з щілиною верхньої губи та альвеолярного відростка, так і ізольовано від них. Вона може бути повного і неповною.
Повна щілина поширюється як на тверде, так і на м’яке піднебіння (наскрізна), неповна може обмежуватись або лише м’яким піднебінням, або м’яким та частиною твердого (частіше), або лише твердим (рідше).

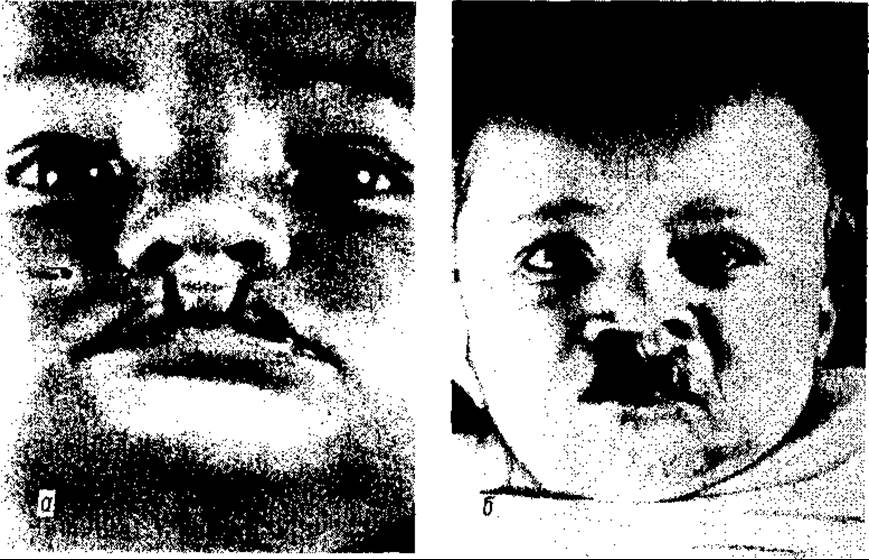
Мал. 117. Однобічне незарощення верхньої губи: а — часткове неповне; б — повне незарощення губи, альвеолярного відростка та піднебіння
Щілини верхньої губи, а також альвеолярного відростка верхньої щелепи та піднебіння пов’язані з ембріональною затримкою росту та зрощення верхньощелепних відростків унаслідок ще недостатньо відомих зовнішніх та внутрішніх впливів на ембріон (у період 8—12-го тижня його розвитку), в тому числі й спадкових. Про роль спадковості свідчить наявність цих вад у сімейному анамнезі хворих. Вважається, що вади спадкового походження передаються за рецесивним типом.
Щілина губи, особливо повна, як однобічна (мал. 117), такі, особливо, двобічна (мал. 118), призводить до зміни крила, кінчика та перегородки носа.
Комбінація щілини губи з щілиною піднебіння супроводжується також і значними анатомічними порушеннями — збільшенням об’єму глотки, порожнини носа, недорозвитком альвеолярного відростка та відростків піднебіння верхньої щелепи, гіпертрофією носових раковин.
Ці вади зумовлюють тяжкі порушення як Харчування, так і Дихання, швидкий розвиток запалення дихальних шляхів, аж до запалення легень, органів слуху, порожнини носа, кишечнику.
Лікування щілин губи та піднебіння проводиться хірургічним шляхом. Ортопедичне лікування щілин твердого піднебіння (обтураторами), яке колись було поширеним, зараз застосовують у поодиноких випадках у дорослих (через переважно тяжкі супутні хвороби).
Пацієнтів з щілинами верхньої губи оперують у перші тижні життя (рідше в перші місяці), подекуди навіть ще до виписування породіллі з пологового будинку.
Хворих з комбінацією щілин губи та піднебіння оперують або в два етапи — в ранньому віці ліквідують лише щілину губи, а дефект піднебіння закривають у 3—6 років, іноді оперують одномоментно, але не раніше одного року після народження.
Дуже рання операція із закриття щілини піднебіння (уранопластика), окрім того, що є складним і тяжким втручанням, несе ризик недорозвитку та деформації верхньої щелепи.
Мал. 118. Двобічне незарощення верхньої губи: а — лише губи; б — губи та щелепи (Ф. Буріан)
Післяопераційна летальність не перевищує 1—2 %.
Пластику дефектів як губи, так і твердого піднебіння здійснюють місцевими м’якими тканинами, що оточують дефект (аутопластика), шляхом їх мобілізації (губи та слизової оболонки і окістя твердого піднебіння) та переміщення і зшивання за різними способами (О. І. Євдокимова, А. А. Лімберга, Ю. Й. Бернадського та ін.).
Хірургічне втручання доповнюють фізіотерапевтичними та логопедичними методами.
ВАДИ ШИЇ
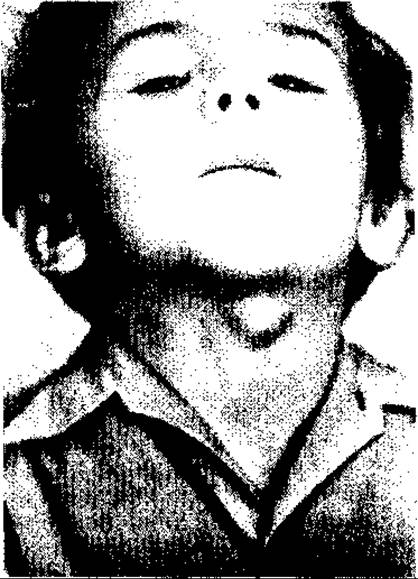
Серединна кіста та серединна фістула шиї. Серединна кіста шиї виникає з незарощеної щитовидно-язикової протоки (ductus thyreoglosus), яка в ембріональний період з’єднує порожнину головного кінця первинної кишки з щитовидною залозою.
Уже в І триместрі ембріона зв’язок щитовидної залози з кишкою розривається, протока закривається, залишаючи на спинці кореня язика ямочку, так званий сліпий отвір (foramen coecum).
У разі порушення процесу редукції та облітерації щитоязикової протоки остання переважно в дистальному (аборальному) відділі, рідко на всій відстані, не заростає, залишається відкритою. Якщо вона не зарощена на всій відстані, то відкривається в ротову порожнину (на язиці). Оскільки протока з середини вистелена багатошаровим епітелієм, здатним до секреції слизу (він походить з кишки), останній поступово накопичується, перетворюючи щілинну порожнину протоки на кульоподібний мішок з фіброзною оболонкою — кісту. Накопичення секрету відбувається особливо прискорено за подразнення епітелію протоки мікрофлорою, яка легко потрапляє в неї через лімфатичні та Кровоносні судини або прямим шляхом з ротоглотки (якщо відкривається в порожнину рота), або через лімфатичні та кровоносні судини при інфекційних захворюваннях ротоглотки та верхніх дихальних шляхів пацієнта.
Мал. 119. Серединна кіста шиї
Інфікування протоки прискорює перетворення її на кісту і нерідко призводить до нагноєння кісти. Гостре нагноєння кісти з переходом запального процесу на м’які тканини шиї, що буває переважно у разі відкритих у порожнину рота проток — кіст, призводить до відкриття кісти на шкіру шиї і перетворення її на норицю. Серединна нориця є інфекційним ускладненням серединної кісти, тобто вторинним проявом, а не первинним.
Серединна кіста має вигляд кульоподібної пухлини від 1,5—2 до З— 4 см у діаметрі, яка розташована по середній лінії шиї (це обумовлено тим, що щитоязикова протока є елементом головного, медіального зачатка щитовидної залози) на відрізку між під’язиковою кісткою та верхнім краєм щитовидної залози, головним чином на рівні верхнього відтинку щитовидного хряща (мал. 119). Вміст кісти — біла прозора слизова рідина або ж рідкий гній у разі інфікування її.
Хоч в основі кісти лежить природжений субстрат, клінічно вона виявляється найчастіше у віці 5—12 (переважно 7) років. Вона може виявлятися як раніше, так і пізніше, навіть у похилому віці (після інфікування ротоглотки чи верхних дихальних шляхів унаслідок активізації секреції залишкових незарощених кишень, тобто синусів, щитоязикової протоки. Кіста має форму кулі, твердо-пружну (рідинний вміст) консистенцію, гладеньку поверхню. Безболісна під час промацування, якщо в ній нема запалення (нагноєння). Вона, як і зоб, зміщується під час ковтання разом із гортанню вгору. Ця особливість відрізняє її від інших (позатиреощних) пухлиноподібних утворень шиї.
У діагностиці її допомагають тонкоголкова пункція (одержання слизової чи гнійної рідини) та сканування з 131І чи 99Тх. Останній накопичується не в кісті, а в зобних вузлах, що розвиваються в пірамідальному відростку щитовидної залози (пірамідальний відросток утворюється з дистальної частини ductus thyreoglosus).
Серединні нориці діагностують як за анамнестичними, так і за фізикальними даними: наявність до появи її кісти та запалення; типова (серединна) локалізація (лише іноді отвір нориці дещо відхиляється від середньої лінії шиї); гнійне виділення з нориці, іноді з запаленням шкіри; фістулографія (з ліпоїдолом чи верографіном).
Зв’язок нориці з ротовою порожниною встановлюють шляхом уведення в норицю метиленового синього чи брильянтового зеленого (з одночасним вкладанням у ротову порожнину марлевого тампона); за наскрізного характеру нориці (наявності зв’язку з роговою порожниною) кольоровий антисептик потрапляє в рот, забарвлюючи марлевий тампон.
Серед серединних кіст близько 6 % є дермоїдні кісти, що походять не з ductus thyreoglosus і клінічно не відрізняються від кіст серединних, але мають інший вміст (сальний детрит).
Лікування серединних кіст та нориць — хірургічне. Видаляють кісту чи норицю разом із центральною частиною під’язикової кістки, через яку проходить верхній кінець щитоязикової протоки (чи нориці). Протоку біля кореня язика перев’язують і нижче лігатуру відрізують.

Мал. 120. Бічні кісти шиї (а, б)
З косметичної точки зору втручання треба робити з дугоподібно поперекового доступу (відкритого вгору) нижче від кісти чи навколо нориці. Серединні дермоїдні кісти видаляють без резекції під’язикової кістки.
Бічні кісти (мал. 120) та фістули шиї походять, найімовірніше, з елементів так званих зябрових чи бронхіогенних випинань (кишень) на первинній глотці. Ці кишені, що є начебто ремінісценцією у людини філогенетичного розвитку тваринного світу, є джерелом формування багатьох органів шиї — елементів слухового апарату, гортані, мигдаликів, підгрудинної залози, прищитовидних залоз тощо. Внутрішні кишені походять з ендодерми. Зовнішні кишені — борозни, що лежать між внутрішніми і спрямовані всередину, утворюються з ектодерми. Кишені ці розділені між собою так званими обтураційними мембранами. За нормального ембріогенезу кишені зникають у перші 2 міс розвитку.
Вважають, що у разі передчасного закриття зсередини внутрішньої кишені утворюються бічні кісти із слизовим вмістом. Тоді у разі такого закриття зовнішньої кишені утворюється дермоїдна кіста.
У деяких випадках в обтураційній мембрані між кишенями виникають так звані перфораційні отвори (за нормального розвитку вони не утворюються), які і складають анатомічний субстрат бічних шийних нориць.
Існують і інші гіпотези походження бічних кіст. Р.І. Венгловський (1909) вважав, що вони утворюються із залишків зобно-глоткової протоки (ductus thymo pharyngeus), яка виникає з II—III зябрових дуг.
Деякі вчені розглядають походження кіст як ваду формування лімфатичних вузлів шиї (Е.С. King, 1972).
Бічна кіста буває переважно однобічною і розташовується в зоні верхньої третини бічної поверхні шиї спереду від грудино-ключично-сосковидного м’яза. Але зустрічаються кісти й. під нижнім краєм цього м’яза. Виникає кіста переважно в підлітковому та юнацькому віці, нерідко — в дорослому. Як і серединних кіст, розвиток бічної кісти стимулюється інфекцією. Про це свідчать не тільки виникнення їх у юнацькому і дорослому віці, а й наявність гнійного вмісту в них. Джерелом їх є, мабуть, переважно II, рідше III, зяброві кишені. Кіста має вигляд пухлини овальної форми, яка на обмеженому відрізку деформує шию, виступаючи над рівнем шкіри на 2—4 см. Пухлина не зміщується під час ковтання. Вона має гладеньку поверхню. Консистенція її коливається від м’якої до твердо-пружної з симптомом ундуляції (балотування). Пальпація її за відсутності гострого запалення безболісна, шкіра над кістою не змінена. Ліфматичні вузли шиї не збільшені, якщо кіста не ускладнена флегмоною. Остання буває нерідко, як правило, має тяжкий перебіг (висока Температура ТІЛА ТА інтоксикація). Кіста залягає глибоко, спереду вона вкрита претиреоїдними м’язами та краєм грудино-ключично-сосковидного. Задня її поверхня лежить на судинно-нервовому стовбурі шиї, безпосередньо на внутрішній яремній вені.
Мал. 121. Природжена м’язова кривошия
Лікування кісти хірургічне — видалення її з косо-поперечного розрізу над кістою завдовжки 8—10 см з перетином шийної фасції по передньому краю грудино-ключично-сосковидного м’яза і відведенням останнього зовні, а претиреоїдних м’язів — усередину.
Нориці бічної поверхні шиї. На відміну від кіст, нориці шиї виявляють одразу після народження. Нориці частіше бувають поодинокими, але виявляють і численні та двобічні кісти (максимально до 6 — по 3 з кожного боку).
Найвище кіста локалізується на мочці вуха або спереду від козелка — вона з’єднує норицю з слуховою трубою і походить з І зябрової кишені (перфораційного отвору її обтураційної мембрани).
Нориці, що походять з II та III зябрових кишень, відкриваються звичайно спереду від грудино-ключично-сосковидного м’яза відповідно на середині та на рівні ключиці нижче за лінію шиї. Але частіше одна нориця локалізується в середній чи нижній частині шиї, тобто джерелом її є II зяброва дуга. Нориці майже завжди наскрізні. Якщо вона розвинулася з II дуги, то вона відкривається в глотці під мигдаликом.
Клінічно нориця виявляється у вигляді невеликого отвору на шкірі, з якого під час натискування виділяється крапля світлої слизової рідини. Шкіра навколо отвору нориці переважно чиста, не запалена. У разі запалення, інфекції у нориці, які бувають набагато рідше, ніж у кістах, виділення мають гнійний характер із запаленням шкіри навколо отвору. Така нориця може бути або не наскрізною, себто закінчуватися в м’яких тканинах біля стінки глотки, або утворюватись унаслідок нагноєння бічної кісти та відкриття її на шкірі. В останньому разі отвір на шкірі буває широким, а шкіра запаленою. Хоч переважно на шкірі нориця має точковий отвір, але підшкірна частина її досить широка — 0,3—0,8 см, має товсту стінку. Вона йде вздовж переднього краю грудино-ключично-сосковидного м’яза спочатку підшкірно, а потім заглиблюється і проходить у сагітально-медіальному напрямку до стінки глотки, або між гілками сонної артерії (нориця з II кишені), або за сонною артерією (зовні її) до стінки глотки.
Лікування нориць хірургічне — видаляють їх з косого або з ступінчастого доступу Проктора, який складається з кількох поперечних (один над одним на відстані 2—3 см) розрізів. Численні нориці, крім тих, що локалізуються на вухах, лікують також оперативно (поетапно).
Перед операцією для уточнення напрямку нориці роблять фістулографію, а на операційному столі в норицю вводять кольоровий антисептик для встановлення зв’язку нориці з порожниною глотки та простеження ходу фістули в тканинах.
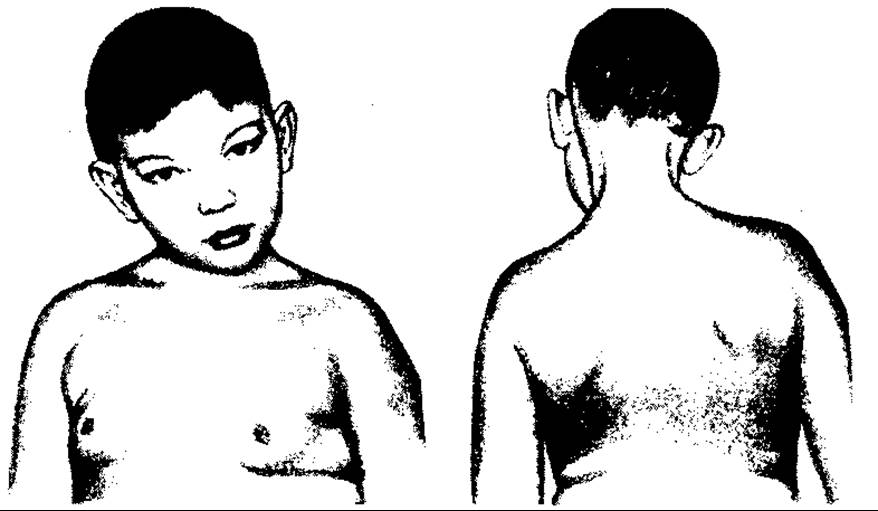
Кривошия (torticolon) — різкий нахил шиї вбік — є вадою різного походження, природжена, так і набута).
Природжена кривошия виникає внаслідок рубцевого переродження і укорочення грудино-ключично-сосковидного м’яза в ембріональний період. Деформація зумовлюється місцевими впливами на плід, неправильним положенням його, можливо, інфекцією. Уражується переважно нижня частина м’яза, яка має вигляд рубцевого тяжа, зв’язаного з власного фасцією шиї та її пластинкою.
Спостерігається головним чином однобічна вада (мал. 121), частіше з правого боку, буває звичайно у дівчаток.
ГОЛОВА дитини нахилена в бік укороченого м’яза та вперед з поворотом обличчя і підборіддя в протилежний бік. При цьому рельєфно виступає натягнутий, як віжка, укорочений м’яз. Нахилена частина обличчя випнута та вкорочена, а протилежна — сплощена, видовжена та дещо атрофована.
Через постійний нахил голови зміщується також плечовий пояс — опущений на боці нахилу, піднятий — на протилежному боці.
У дитини звужене поле зору. За тривалого існування кривошиї з’являється також викривлення хребетного стовпа, себто м’язова кривошия доповнюється кістковою. Діагностика природженої кривошиї у разі вираженої форми її нескладна.
Невеликі ступені її в ранньому віці лікують консервативно — ортопедичним методом, застарілі форми та такі, що не піддаються консервативному лікуванню, потребують хірургічного втручання (повністю видаляють рубцево-перероджену частину м’яза разом із рубцево-зміненими та спаяними з ним фасціями). Корекція шиї і голови відбувається уже на операційному столі. Подальша фізіотерапія завершує реабілітацію.
Гігрома шиї — переважно багатопорожнинна кіста або лімфома. Це одна з найчастіших природжених вад. Головним чином локалізується на шиї, хоч зустрічається також у пахвинній та паховій ділянках. Причина її — порушення розвитку лімфатичної системи.
Анатомічно виявляється утворенням замкнутих багатокамерних лімфатичних мішків, заповнених жовтуватою чи навіть геморагічною серозною рідиною. Відсутність природного відпливу призводить до швидкого накопичення великої кількості рідини сецернуючим епітелієм оболонок лімфоми і її збільшення. Гігрома іноді досягає розмірів голови немовляти і може призвести до асфіксії. Діагноз встановлюють за допомогою огляду. Виявляють кулясте утворення з розстягнутою над ним шкірою та симптомами ундуляції. Лікування оперативне (видалення гігроми).
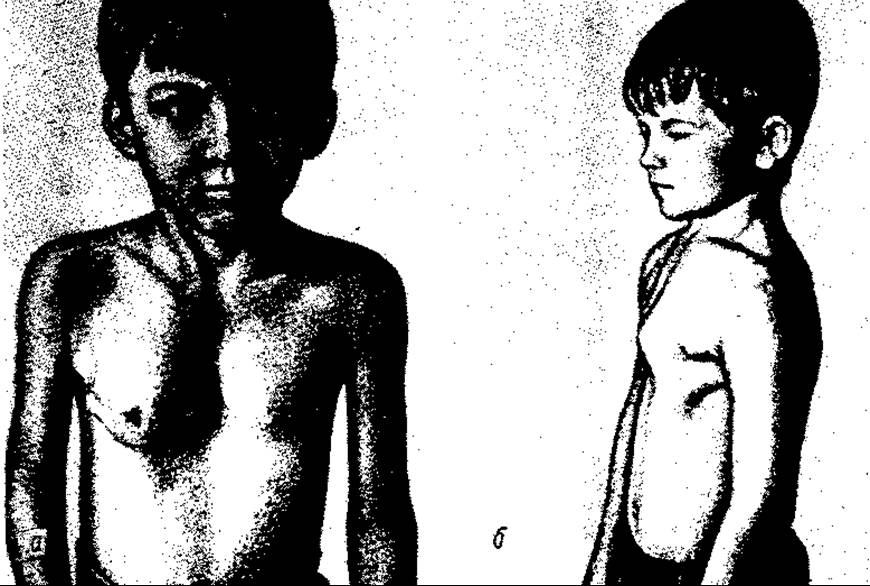
Мал. 122. Деформація груднини: а — лійкоподібна; б — кілеподібна (куряча)
У разі поширення її на обличчя або у грудну клітку видаляють основну частину гігроми, а інші — розтинають, запобігаючи накопиченню рідини та стисненню органів. Радикальне видалення звичайно неможливе в такому випадку. Для зменшення тиску на дихальні шляхи (запобігання асфіксії) пунктують гігрому і відсмоктують з неї рідину. При багатокамерній гігромі пунктують окремо кілька камер.
ВАДИ РОЗВИТКУ В ДІЛЯНЦІ ГРУДНОЇ КЛІТКИ
У лікарській практиці зустрічається чимало вад, що стосуються скелета, грудної клітки, молочної залози та органів дихання.
Лійкоподібна деформація грудної клітки (pectus excavatus) — широке западання тіла груднини з реберними хрящами та мечовидного відростка (мал. 122, а), що деформує передню стінку клітки. Часто комбінується з кіфозом. Окрім косметичного дефекту, вада негативно впливає на Серце — зміщує його і може спричинити порушення його функції, а також функції легенів. Причини розвитку невідомі. Дехто вважає ваду наслідком д испропорції темпів росту діафрагми та груднини. Лікування в тяжких випадках хірургічне — пластична операція.
Кілеподібна (мал. 122, б) деформація грудної клітки (pectus carinatum), або курячі чи голуб’ячі груди (англ. pigeon breast), полягає в кілеподібному випинанні груднини з хрящами ребер вперед. Лікування — пластична операція.

Мал. 123. Додаткова молочна залоза (C.D. Haagensen)
Відсутність розвитку однієї чи обох молочних залоз — амастія — буває дуже рідко. Частіше спостерігаються юнацька гіпертрофія молочних залоз, розвиток додаткової молочної (однієї чи двох, тобто з обох боків) залози (мал. 123), яка локалізується в передній частині пахвинної ямки (політелія). Соски розташовані вздовж так званих молочних ліній, тобто від пахви до лобкового симфізу.
Лікування додаткових молочних залоз — оперативне видалення їх. У деяких випадках при гіпертрофії молочних залоз проводять також пластичну операцію за косметичними показаннями. Політелія не потребує лікування.
Рідкісною вадою грудної клітки є розщеплення груднини. За легкої форми вади виявляють невелику поздовжню щілину, тяжкі ж форми характеризуються широким розходженням сегментів груднини та шкіри з випаданням через дефект серця з перикардом. Цю форму вади лікують за допомогою пластичної операції.
Ще складнішою є так звана дистальна фісура, яка звичайно поєднується з вадою діафрагми та черевної стінки (омфалоцеле). Для лікування її потрібна дуже складна операція.
ВАДИ РОЗВИТКУ ЧЕРЕВНОЇ СТІНКИ
Найбільш відомі та поширені природжені вади черевної стінки — це ембріональна грижа, чи омфалоцеле (omphalocele), пуповинна грижа, грижа пупкового канатика, грижа пупкова та пахово-мошонкова грижа.
Найтяжчою вадою є омфалоцеле, чи евентрація внутрішніх органів черевної положнини через великий дефект у черевній стінці в зародкову амніотичну оболонку в ділянці пупкового канатика.
У період між 6-м та 10-м тижнями внутрішньоутробного розвитку целомічна порожнина зміщується в бік пупкового тяжа (канатика), куди й спрямовуються передусім кишкові петлі. Останні ростуть швидше, ніж збільшується сама порожнина. Після 10-го тижня Черевна порожнина (її стінка) росте швидше і внутрішні органи занурюються в неї. За десинхронізації між цими процесами може створитись ситуація, коли стінка черевної порожнини (її тканини та шкіра) затримується в своєму розвитку, в стінці (в центральній частині її) створюється дефект, через який випадають органи черевної порожнини. Вони вкриті лише зрощеними між собою очеревиною та амніотичною оболонкою, яка має товщину до 1 мм і прозорий драглистий вигляд.
Другий варіант природженої черевної грижі — грижа, що виходить через пупкове кільце між дефектом у прямих м’язах за нормально розвиненої черевної стінки. Пупкова природжена грижа вкрита лише шкірою. Ця грижа невелика за розмірами і за перебігом докорінно відрізняється від омфалоцеле. Шкіра захищає черевну порожнину від проникнення інфекції, тоді як омфалоцеле вкрите лише прозорою драглистою оболонкою, через яку швидко проникає інфекція і призводить до перитоніту. Тому хірургічне втручання у разі омфалоцеле треба виконувати зразу після народження дитини.
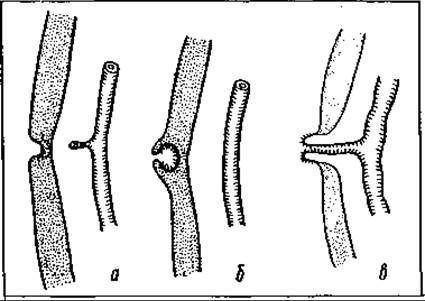
Мал. 124. Види розвитку пупково-кишкової протоки — ductus omphaloentericus: а — дивертикул Меккеля; б — сліпа нориця; в — повне незарощення пупковомезентеріальної протоки
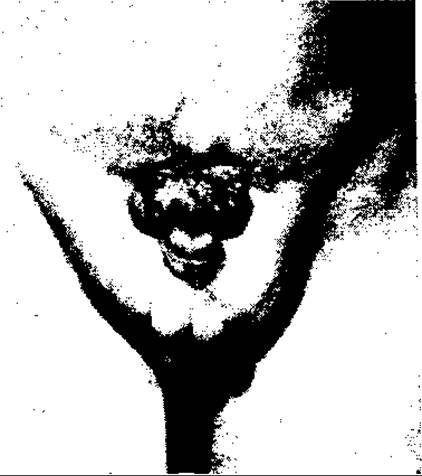
Мал. 125. Екстрофія сечового міхура з епіспадією
За великих розмірів омфалоцеле проводять нерадикальну операцію (вкривають грижу мобілізованою шкірою). Пупкова грижа може бути оперована пізніше, після спроб консервативного лікування.
Природжена пахова, або пахово-мошонкова, грижа патогенетично зв’язана з процесом опускання яєчок (descendes testiculorum) з черевної порожнини в пахову ділянку. Цьому процесу передує утворення так званого піхвового відростка очеревини (processus vaginalis), в який опускається Яєчко (останнє інвагінується в ньому разом із задньою стінкою). Після опускання Яєчка, яке відбувається перед народженням, порожнина відростка очеревини над ним і вгорі до самого отвору облітерується і закривається.
Якщо очеревинний відросток не закривається до пологів, після останніх він під впливом підвищення внутрішньочеревного тиску (який збільшується під час напруження м’язів дитини, особливо плачу та крику) заповнюється внутрішніми органами, насамперед кишками, і перетворюється на грижу. Ознака її — виявлення органів черевної порожнини в грижовому мішку разом із яєчком. Оскільки така грижа часто защемлюється, лікують її оперативним шляхом у ранньому віці.
У ділянці пупкової ямки можуть, окрім гриж, спостерігатись вади, що пов’язані з неповним зворотним розвитком ембріональних пупково-мезентеріальних та пупково-сечоміхурових проток.
У разі повного їх незакриття в пупковій. ямці відкривається звичайно отвір: кишкова нориця (мал. 124) — за незакритої пупково-мезентеріальної протоки та сечова — за незакриття пупково-сечоміхурової протоки.
Якщо незакрита лише дистальна частина проток, в пупковій ямці відкриваються сліпі синуси цих проток або лише частина слизової оболонки мезентеріальної протоки (так званий пупковий поліп), яка секретує слиз. Ці не повністю облітеровані протоки бувають джерелом розвитку абсцесів черевної стінки та пупка. Лікування цих вад — хірургічне.
Серед зовнішніх вад сечовидільної системи найтяжчою є екстрофія сечового міхура (мал. 125). Вада полягає в недорозвитку передньої стінки сечового міхура та передньої стінки черевної порожнини над ним з розділенням кісток лобкового симфізу і випаданням через дефект черевної стінки слизової оболонки міхура, нерідко з вхідними отворами сечоводів на її поверхні.
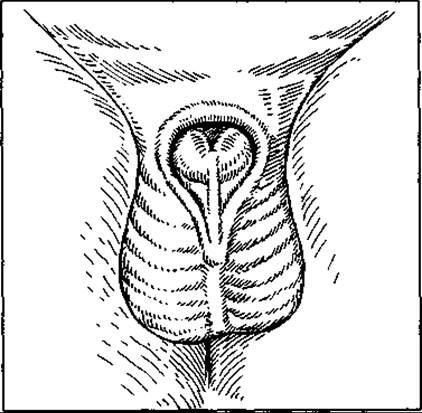
Maл. 126. Епіспадія
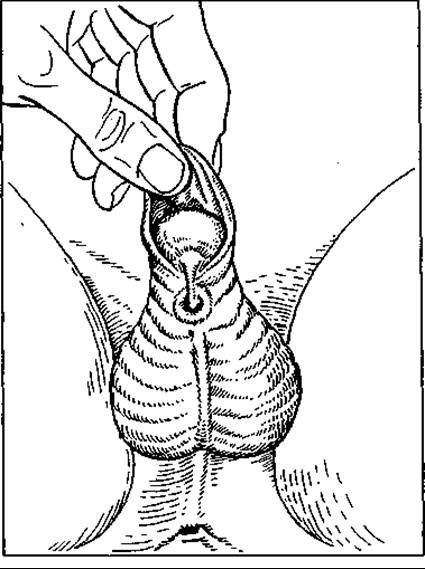
Мал. 127. Гіпоспадія
Діти з такою вадою через порушення з’єднання лобкових кісток ходять перевалюючись («качача» хода). Ця вада часто комбінується з іншими дефектами, зокрема, неопусканням яєчок тощо.
Екстрофія зустрічається з частотою 1 на 100—120 тис. хлопчиків і 1 на 500 тис. дівчаток.
Вада супроводжується постійним виділенням сечі на шкіру, мацерацією її та запаленням.
Лікування вади полягає в складній пластичній операції сечового міхура та передньої стінки живота, а у разі неможливості усунення вади таким шляхом застосовують пересадку стінки міхура з отворами сечоводів у кишку.
Серед вад сечівника розрізняють ектопічне розташування отвору каналу на передній (верхній) поверхні статевого члена (епіспадія) або на нижній (гіпоспадія).
Епіспадія зустрічається рідко, але належить до дуже тяжких патологій — якщо її неможливо усунути (мал. 126). Найлегший її тип — головковий, чи баланітний, за якого отвір сечівника відкривається на головці статевого члена, Нетримання сечі при цьому типі немає. Другий тип її — стовбуровий, чи корпорний, коли Сечівник відкривається вздовж усієї верхньої поверхні статевого члена. При цьому типі нетримання сечі може бути, а може не бути, якщо нема недостатності сфінктера шийки сечового міхура. Третій тип спостерігається найчастіше (penopubicum). Вада охоплює всю довжину статевого члена та поширюється на невелику відстань черевної стінки. Супроводжується незростанням лобкових кісток.
Епіспадія зустрічається переважно у хлопчиків, але може бути також і в дівчаток (кліторний, промежинний і повний тип, який супроводжується відокремленням сечівника, сфінктера, сечового міхура). Операція перспективніша в перших двох випадках, у третьому потрібна пересадка сечоводів (у віці 3—6 років).
Гіпоспадія (мал. 127) зустрічається значно частіше, ніж епіспадія (у 20 разів), і буває трьох типів: френулярна, чи вуздечкова, корпорна, або середньої частини тіла статевого члена, і пеноскротальна, або промежинна. У останньому випадку визначити стать дитини, бо зовнішні ознаки не чітко виражені. Гіпоспадія завжди комбінується з різними деформаціями — викривленням статевого члена, рудиментарним статевим членом. Лікування оперативне.
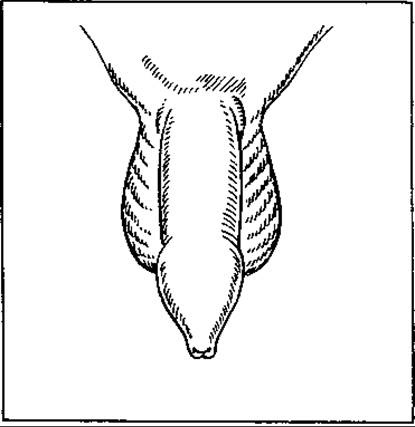
Мал. 128. Фімоз
Фімоз — вада, що полягає в різкому звуженні отвору крайньої плоті статевого члена (мал. 128). Під нею (між нею і головкою) накопичуються сеча та смегма, що призводить до подразнення та запалення як плоті, так (часто) і головки члена, тобто виникає постит та баланіт. У дорослому віці ця вада може порушувати статеву функцію. Насильне зсовування плоті над головкою може спричинити защемлення головки, тобто парафімоз. Лікування фімозу хірургічне — видалення дистальної частини крайньої плоті, де її отвір широкий.
Вади розвитку задньопрохідного отвору та прямої кишки
Вади розвитку цих відрізків травного каналу найпоширеніші. Зустрічаються дещо частіше у хлопчиків. Найтяжчі серед них атрезія задньо-прохідного отвору (atresia ani) та прямої кишки (atresia recti) і агангліоз ректосигмоїдного відділу товстої кишки, або ж хвороба Гіршпрунга. Атрезія задньопрохідного отвору полягає у відсутності отвору в пряму кишку (він закритий шкірою) через затримку в ембріогенезі формування перфорації анальної мембрани (заслінки). Атрезія прямої кишки — це сліпе закінчення її вище від анального отвору.
У ранньому ембріогенезі, коли ембріон має довжину 7,5 мм, у нього формується клоака — загальна порожнина, що об’єднує сечостатевий синус та задню кишку.
З часом ця порожнина ділиться сечогенітальною та анальною мембранами на порожнини.
Внаслідок затримки чи порушення відокремлення сечогенітального синуса від порожнини кишки мембранами та перфорації останніх формуються різні вади в ділянці органів (мал. 129), а саме: 1) стеноз анального отвору (отвір є, але він різко звужений); 2) анальний отвір відсутній через відсутність перфорації його мембрани; 3) відсутність анального отвору та сліпе закінчення прямої кишки на деякій відстані від закритого анального отвору; 4) анальний отвір і анальний канал розвинені нормально, але вище від нього пряма кишка являє собою сліпий мішок, тобто між нею і анальним каналом є перегородка.
Вади перших трьох типів діагностуються у першу добу, тоді як четвертий — лише через кілька діб, внаслідок гострої кишкової непрохідності. Тому наслідки лікування цього типу завжди гірші. У 70 % дітей з цими вадами існують нориці (фістули) прямої кишки в сечостатеву систему чи в промежину.
Лікування цих вад — лише хірургічне. Найпростішим є втручання при другому типі вад. Воно зводиться до розтину шкіри над анальним кільцем. При 3-му та 4-му типах дефекту потрібні операції накладання сигмо- стоми з радикальною операцією у віці до року.
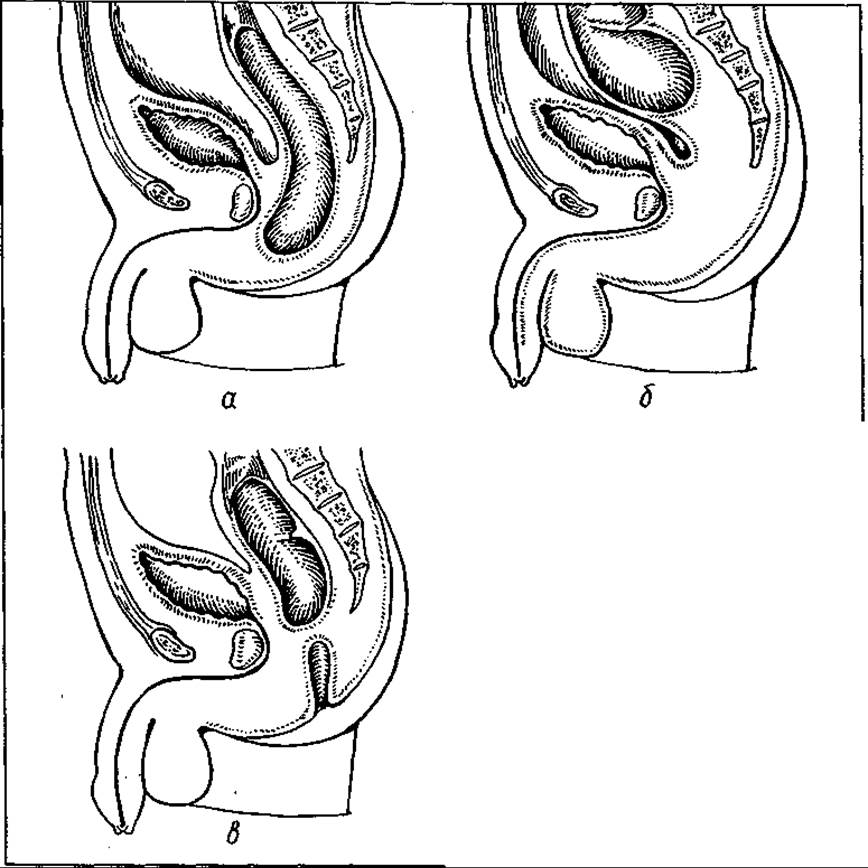
Мая. 129. Атрезія прямої кишки: а — атрезія анального отвору; б — атрезія анального отвору та прямої кишки; в — атрезія власне прямої кишки
ВАДИ РОЗВИТКУ СІДНИЦЬ, ПРОМЕЖИНИ ТА ЧОЛОВІЧИХ ЗОВНІШНІХ СТАТЕВИХ ОРГАНІВ
У ділянці сідниць, крижово-куприковій та промежини зустрічаються дермоїдні кісти, нориці, тератома, додаткова мошонка, неопущені яєчка (на промежині) тощо.
Природжені нориці є головним чином компонентом вади розвитку прямої кишки, її атрезії та сполучення її порожнини з навколишнім середовищем (з поверхнею шкіри) фістулою.
Дермоїдні кісти локалізуються переважно попереду та дещо нижче від куприка, тератома — в ділянці крижів, куприка та промежини. Вони різко виступають над поверхнею шкіри або поширюються вглиб тканин. Дермоїдні кісти містять елементи придатків шкіри (жироподібні маси та Волосся). Тератома, окрім придатків шкіри, містить ще елементи інших, крім ектодерми, зародкових тканин, зокрема ентодерми (найчастіше кісткову або хрящову тканину).
Крипторхізм (гр. — ciyptos — прихований; orxos — яєчко) — неопущення яєчка в мошонку із затримкою його на різних етапах опускання з черевної порожнини в мошонку.
Вада може бути одно- та двобічною. Якщо яєчка затримуються в черевній порожнині, тобто їх немає в паховому каналі, вада називається анорхізмом (за відсутності одного яєчка — монорхізм). У разі затримки яєчка в паховому каналі або за межами мошонки під шкірою розвивається крипторхізм. Здебільшого буває монорхізм чи крипторхізм. Неопущене яєчко може бути на різних рівнях в паховому каналі або за його зовнішнім кільцем, але воно фіксується над апоневрозом зовнішнього косого м’яза спайками.
Яєчко, як правило, гіпоплазоване. Лікування вади оперативне. За внутрішньочеревного розташування його потрібна оперативна ревізія черевної порожнини.
Якщо яєчко гіпоплазоване значною мірою, його видаляють, оскільки воно може перероджуватись на злоякісну пухлину.
При паховому крипторхізмі яєчко опускають оперативним шляхом у мошонку. Операцію треба виконувати у віці 3—4 роки, оскільки пізніше в яєчку спостерігаються необоротні дистрофічні зміни.
ВАДИ ЖІНОЧИХ ЗОВНІШНИХ СТАТЕВИХ ОРГАНІВ
Серед вад цих органів найчастіше спостерігаються неперфорована дівоча пліва піхви (hymen feminimus imperforatus) та несправжній жіночий гермафродитизм.
Неперфорована пліва, що закриває порожнину матки та частково піхви, спричиняє затримку та накопичення менструальної крові в порожнині матки та піхви (гематокольпос) у маткових трубах і навіть у черевній порожнині. Різко розтягує та збільшує матку (мегаметрум) та випинає hymen донизу. Ця вада призводить до запалення матки, навіть до перитоніту.
Потрібні рання діагностика (кольпоскопія) і розтин пліви. До тяжких вад жіночих статевих органів належить несправжній гермафродитизм, зумовлений переважно природженою наднирковою гіперплазією (наднирковими андрогенами). Він супроводжується недорозвитком піхви та матки і гіпертрофією клітора (мал. 130).
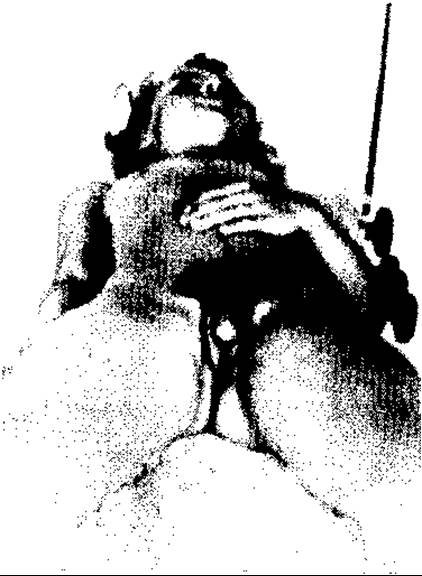
Мал. 130. Жіночий псевдогермафродитизм з кліторомегалією
Вада дуже складна і вимагає точної діагностики як генетичної (хромосомної) статі, так і характеру внутрішніх статевих залоз. Лише після цього може бути проведене лікування із застосуванням хірургічних коригуючих операцій.
Жіночий несправжній гермафродитизм може поєднуватись з іншими вадами сечостатевої системи та прямої кишки.
ВАДИ КІНЦІВОК
Вади кінцівок численні та різноманітні — від незначних до тяжких каліцтв. Нерідко зустрічається зрощення пальців — синдактилія, збільшення кількості пальців — полідактилія (мал. 131) або зменшення її — олігодакзилія, додаткова кінцева (нігтьова) фаланга (мал. 132), а також недорозвиток пальців та деформації кистей і стоп.
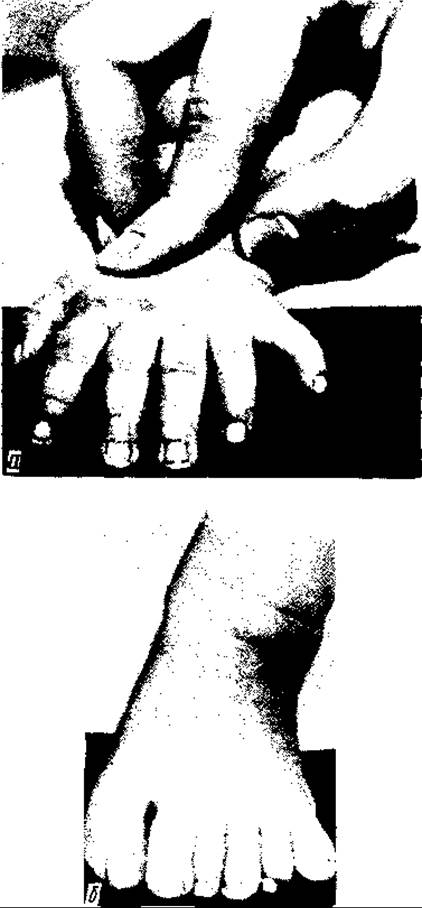
Мал. 131. Полідактилія верхньої (а) та нижньої (б) кінцівок
До рідкісних аномалій належать відсутність усіх кінцівок — амелія, нижніх (apus) чи верхніх кінцівок (abrachius); зрощення нижніх кінцівок (simpus або sirena), відсутність кісток плеча та передпліччя, відсутність стегон і гомілок і прикріплення кистей або стоп до плечового чи тазового поясів (фокомелія).
Зустрічаються також недорозвиненість окремих кісток чи сегментів кінцівок, збільшення розмірів окремих пальців — Макродактилія, природжені деформації кінцівок у формі перетяжок, природжені види слоновості тощо. Відносно часто спостерігаються природжений вивих стегна та клишоногість.
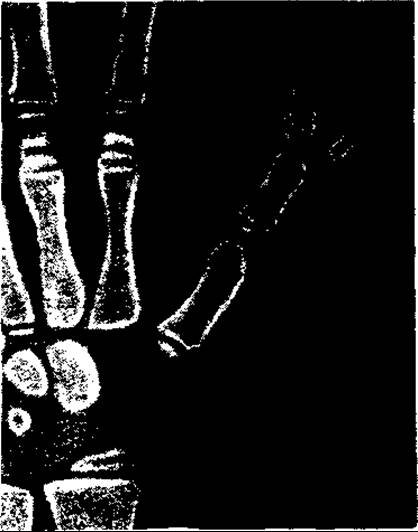
Мал. 132. Додактова фаланга І пальця верхніх кінцівок
Вивих стегна зустрічається переважно у дівчаток. Ознаками його є такі специфічні симптоми: дитина починає набагато пізніше ходити і ходить, як качка; симптом Тренделенбурга (сіднична складка на боці вивиху розташовується вище, ніж на здоровій половині (мал. 133), обмежені відведення і ротація в латеральний бік.
Лікування в дошкільному дитячому віці — ортопедичне, пізніше — оперативне.
Клишоногість (pes equinovarus) — природжена вада стопи, яка буває переважно у хлопчиків і зумовлюється порушенням розвитку нервової системи чи м’язів, а також аномаліями розвитку оболонок плода. Стопа вивернута в середину, підошовною поверхнею вгору (супінована), зігнута в бік підошви, зв’язок кісток стопи з кістками гомілки порушений. М’язи гомілки атрофічні, стегна конічні, хода незграбна. Лікування — ортопедично-хірургічне.
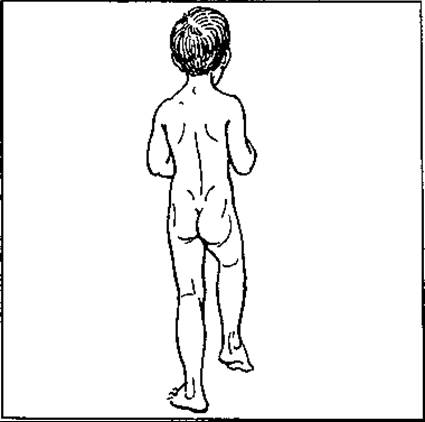
Мал. 133. Природжений вивих стегна — синдром Тренделенбурга
ВАДИ РОЗВИТКУ ВНУТРІШНІХ ОРГАНІВ ТА ТКАНИН
Вади розвитку внутрішніх органів та тканин, зовнішньо не виявляючись, супроводжуються різними функціональними порушеннями, які виявляють або відразу після народження, або на різних етапах життя.
Деякі вади можуть бути несумісними з життям взагалі або смертельними без термінової хірургічної корекції. Інколи вади можуть не спричинювати порушень в організмі хворого і виявлятись випадково чи в разі розвитку якихось тяжких ускладнень.
ВАДИ ТРАВНОГО КАНАЛУ ТА ОРГАНІВ ЧЕРЕВНОЇ ПОРОЖНИНИ
Вади розвитку стравоходу. Найчастішими вадами стравоходу є стеноз та атрезія його. За анатомічною формою зустрічається 6—7 типів дефектів. Серед них розрізняють вади власне стравоходу (рідко) та в комбінації з вадами трахеї, наявністю зв’язку (нориці) недорозвиненого чи розвиненого стравоходу з порожниною трахеї (переважно).
Власне серед вад стравоходу треба виділити передусім стеноз його та атрезію. Стеноз — це вада, що полягає в значному звуженні стравоходу, головним чином у середній третині його. Це найлегша вада. За значного ступеня стенозу вона виявляється у перші тижні або місяці життя і пізніше, коли дитині починають давати напіврідку чи густу їжу.
Рідкісна форма чистої вади стравоходу — це атрезія його на рівні верхньої і середньої третин без зв’язку з трахеєю. Вада виявляється в перші 2 доби після народження і без хірургічної допомоги несумісна з життям.
Всі інші форми атрезії стравоходу поєднуються з вадами трахеї. За формою розрізняють до 4—5 типів таких вад. Серед них найчастішою (90 %) є атрезія верхнього кінця стравоходу в поєднанні із сполученням другого, дистального, відрізка стравоходу своїм верхнім кінцем з трахеєю вище від її біфуркації. Ця вада, як інші, що супроводжуються норицями з трахеєю, клінічно виявляється в перші 2 доби після народження (надмірне слиновиділення та гіперпродукція слини, яка не надходить з порожнини рота в Стравохід через його непрохідність, кашель, посиніння та широко розплющені очі під час ссання грудей).
Діагноз уточнюють за допомогою рентгенографії стравоходу (в останній через катетер уводять 2 мл ліпоїдолу). Лікування цієї тяжкої вади — хірургічне. Без нього діти гинуть від легеневих ускладнень (пневмонії) та неможливості харчування.
До 1939 р. всі способи хірургічного лікування вад були безуспішними. Зараз багатьом пацієнтам вдається провести езофагопластику та ліквідувати норицю трахеї. Післяопераційна смертність становить 30— 40 %. Атрезія стравоходу часто комбінується з вадами інших органів, особливо вадами серця та ректоанальної зони.
У нижньому кінці стравоходу спостерігаються такі вади, як кардіостеноз — звуження кардіальної частини стравоходу (її лікують дилатацією, розширенням стравоходу механічним шляхом бужами або ж пневмобалонами), а також халазія стравоходу, тобто розслаблення, зяяння його кардіальної частини з випаданням слизової оболонки в порожнину шлунка.
ВАДИ РОЗВИТКУ ШЛУНКА ТА КИШЕЧНИКУ
Пілороспазм та гіпертрофічний пілоростеноз. Ці природжені вади розвитку полягають відповідно у патологічному спазмові воротаря шлунка та в гіпертрофії гладенького м’яза його жому. Вади розвиваються переважно у первістків і можуть бути сімейними.
Клінічна картина їх майже тотожна і виявляється симптомами високої непрохідності травного каналу (порушення обміну речовин, насамперед водно-електролітного, та харчування). Блювання з’являється переважно через 10 діб і швидко призводить до зневоднення та втрати маси тіла. Лікування головним чином оперативне.
При пілороспазмі вздовж розтинають м’язовий шар та пілоричний жом шлунка до слизової оболонки. При пілоростенозі роблять наскрізний розріз стінки шлунка вздовж у пілоричному відділі зі зрізуванням гіпертрофованого м’язового жому.
Серед вад тонкої кишки спостерігаються стеноз дванадцятипалої та клубової кишок (рідко), подвоєння різних сегментів її, так званий дивертикул Меккеля. Звуження дванадцятипалої кишки може бути як самостійним (рідко), так і розвиватися внаслідок так званої кільцеподібної підшлункової залози, яка стискає дванадцятипалу кишку.
Дивертикул Меккеля — це бічний відросток (різної довжини) тонкої кишки (клубової) на відстані 50—70 см проксимальніше від переходу її в сліпу. Він є залишком необлітерованої жовткової ембріональної протоки.
Коли жовткова протока не заростає, вона перетворюється на кишкову норицю, що відкривається в ямці пупка.
Діагностика дивертикула Меккеля можлива за допомогою рентгенологічного дослідження тонкої кишки з приводу інших причин, оскільки він не зумовлює порушень до появи в ньому запалення — дивертикуліту. Останній має такий самий перебіг, як апендицит, і дивертикул звичайно виявляють під час апендектомії.
Природжений мегаколон, чи хвороба Гіршпрунга — аномалія розвитку проктосигмоїдального відділу товстої кишки, яка полягає в різкому звуженні та рубцевих змінах, а головне — в пригніченні перистальтики в цій ділянці.
Вада пов’язана з дефіцитом або навіть з відсутністю гангліозних клітин інтрамурального нервового сплетення в стінці цього сегмента товстої кишки, через що її ще називають агангліозом товстої кишки.
Різкі функціонально-анатомічні зміни в проктосигмоїдальному відділі призводять до хронічної непрохідності, гіпертрофії стінки товстої кишки вище від рівня непрохідності з подальшою дилатацією кишки та затримкою фекальних мас у кишечнику. Сигмовидна та низхідна кишки, а часто й усі інші відрізки товстої кишки дуже розширені (їх діаметр досягає 15—20 см) і переповнені злежаними каловими масами.
У разі природженого мегаколону діти відстають у розвитку, виснажені. Живіт різко збільшений, м’язи нерозвинені (мал. 134).
Лікування вади оперативне — видалення проктосигмоїдальної агангліозної ділянки та різко зміненого сегмента сигмовидної кишки із зведенням на промежину дистального кінця товстої кишки. Операція ця проводиться в кількох модифікаціях, але всі вони виконуються зі збереженням сфінктера прямої кишки.
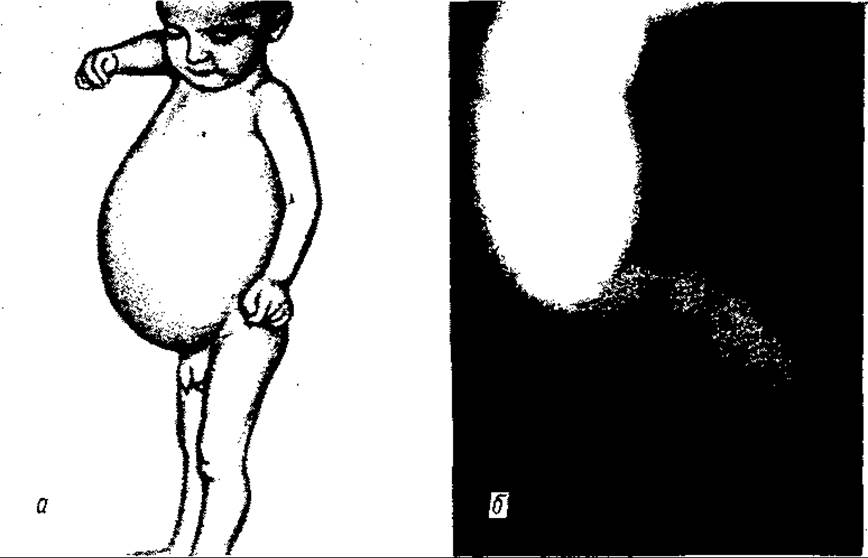
Мал. 134. Хвороба Пршпрунга:
а — загальний вигляд хворого; б — видалений препарат прямої та сигмовидної ободової кишок (М.Б. Сітковський)
ВАДИ ЖОВЧНИХ ШЛЯХІВ ТА ПЕЧІНКИ
Серед вад жовчних шляхів та печінки зустрічаються атрезія загальної жовчної протоки, агенезія її, подвоєння жовчного міхура, прості кісти печінки та агенезія лівої частки печінки.
Найтяжчою вадою є атрезія жовчної протоки, яка супроводжується жовтяницею і без оперативного втручання несумісна з життям.
ВАДИ ПОЛОЖЕННЯ ОРГАНІВ ЧЕРЕВНОЇ ПОРОЖНИНИ, ФОРМОТВОРЕННЯ ОЧЕРЕВИНИ ТА САЛЬНИКА
До вад органів черевної порожнини належить незвичне їх положення, так зване зворотне (сліпа кишка з червоподібним відростком лежить зліва, а сигмовидна — справа тощо). Іноді воно супроводжується загальним зворотним положенням внутрішніх органів (situs viscerum inversus), у тому числі серця (декстрокардія).
Вади формотворення очеревини та сальника полягають переважно в утворенні очеревиною великих кишень у ділянці переходу її з органа на орган чи з черевної стінки на орган та наявності отворів у очеревині й сальнику. Ці вади в більшості випадків не порушують функції органів черевної порожнини, але інколи стають причиною гострої та хронічної непрохідності внаслідок защемлення в цих кишенях та отворах кишкових петель та сальника.
Найчастіше вади очеревини локалізуються в ділянках дуоденального переходу тонкої кишки, де вона утворює глибоку кишеню, в якій нерідко защемлюються кишкові петлі (грижа трейтцевої зв’язки), в зоні переходу клубової кишки в сліпу (клубово-сліпокишкова ямка), а також у передній стінці очеревини над сечовим міхуром, де вона утворює надміхурові ямочки, в яких може бути защемлення органів.
«Вікна» частіше спостерігаються в брижах кишечнику та в сальнику (в них також можливе защемлення кишкових петель).
Рідкісною вадою є подвоєння очеревини (весь кишечник лежить у замкненому очеревинному мішку в середині нормально сформованої черевної порожнини).
ВАДИ СИСТЕМИ СЕЧОВИДІЛЕННЯ
Це численні вади, починаючи від незначних до тяжких. Серед вад нирок зустрічаються недорозвиток (гіпоплазія) однієї чи обох нирок, їх ектопія (дистопія — тазова, здухвинна), зрощення нирок між собою полюсами, частіше нижніми (Підковоподібна Нирка), подвоєння нирки чи сечоводів, Полікістоз нирок тощо.
Багато з цих вад сприяють розвитку запальних та дистрофічних процесів, зокрема пієліту, пієлонефриту, сечокам’яної хвороби.
Подвоєний Сечовід, який іноді може впадати не в Сечовий міхур, а в сечівник, спричинює інконтиненцію (нетримання) сечі. Частішими, однак, є вади нижніх відділів цієї системи, зокрема зовнішні вади сечового міхура та сечівника, про які йшлося раніше.
ВАДИ ВНУТРІШНІХ ЖІНОЧИХ СТАТЕВИХ ОРГАНІВ
Велика кількість вад спостерігається у внутрішніх жіночих статевих органах. У разі дефектів злиття мюллерової протоки можливе подвоєння матки, труб та піхви. Поширені вади за типом гіпоплазії, аж до аплазії матки, піхви, яєчників чи маткових труб.
У деяких хворих з вторинними жіночими статевими ознаками замість яєчника виявляють яєчко (ovotestis). Якщо воно міститься в пахвинних ділянках, його видаляють, оскільки воно часто малігнізується.
ВАДИ СИСТЕМИ ДИХАННЯ ТА ДІАФРАГМИ
В легенях зустрічаються такі вдай: агенезія Легені чи частки її; розвиток додаткової частки з бронхом; неправильний поділ легень на частки; природжені бронхоектази та полікістоз легень. Лікування проводять лише за ускладнення останніх вад (нагнійні процеси).
У дихальних шляхах зустрічаються такі вади, як атрезія та Стеноз гортані чи трахеї, які несумісні з життям або вимагають (стеноз) хірургічного лікування.
До тяжких внутрішніх вад належить діафрагмальна грижа — наявність отвору в грудочеревній перетинці різної величини, аж до повної відсутності діафрагми.
Вада супроводжується порушенням функції як серцево-судинної і дихальної систем (серцебиття, задишка, гіпоксемія), так і органів травлення внаслідок випадання їх (шлунка, кишок) у плевральну порожнину.
За вузьких отворів у діафрагмі в них можливі защемлення шлунка, кишок і розвиток гострої їх непрохідності. Природжені грижі діафрагми іноді поєднуються з іншими вадами, особливо зовнішніми (фісурою грудної стінки, щілиною губи та піднебіння тощо). Лікування діафрагмальних гриж оперативне.
ВАДИ СЕРЦЯ
Вади серця спостерігаються у 0,6— 0,8% новонароджених. Нараховується до 100 видів вад серця. Це вади власне серця та судинних стовбурів.
Найчастіше спостерігаються відкрита артеріальна протока, стеноз легеневої артерії, коарктація аорта, дефекти міжпередсердних та міжшлуночкових перегородок (так звані септальні вади) та комбінована Вада серця і магістральних судин (тетрада Фалло).
Відкрита артеріальна протока. Вада полягає в збереженні незакритою після народження дитини протоки між аортою та легеневою артерією (боталової протоки). На її частку припадає до 15—20 % усіх вад серця.
Вада клінічно починає виявлятись переважно в шкільному віці. З’являються задишка під час виконання фізичних вправ, відставання в рості, мала резистентність до інфекції тощо. Дефект легко діагностується фізикально.
Головною ознакою його є голосний систолодіастолічний (так званий машинний) шум у серці над усією його поверхнею, особливо в другому міжребер’ї зліва від груднини. Серце збільшене помірно. При рентгенографічному дослідженні спостерігаються розширення легеневого стовбура та «танець» кореня.
Вада призводить до підвищення тиску в легеневій артерії, гіпертензії в малому колі кровообігу, гіпертрофії його судин та гіпертрофії і розширення правого шлуночка (синдром Ейзенменгера). Згодом тиск у легеневій артерії може стати таким самим, як і в аорті, а потім навіть більшим — через збільшення легеневої гіпертензії та ослаблення серцевого м’яза. З поверненням кровообігу справа (з легеневої артерії) вліво (аорту) розвивається недостатність з подальшим смертельним кінцем у юнацькому віці. За умови ранньої діагностики цю ваду виліковують хірургічним шляхом — перев’язують протоку з двох кінців та інколи перерізують її. Летальність становить до 1 %. Ця операція стала однією з найпростіших в кардіохірургії. Перша така операція була зроблена в 1938 р.
Коарктація аорти — вада низхідної частини дуги аорти, що полягає в звуженні її (мал. 135). Характеризується різким підвищенням систолічного тиску у верхніх відділах тіла (голова, груди, верхні кінцівки) і посиленням кровопостачання цієї частини тіла та різким зниженням тиску і кровопостачання в нижньому відділі тіла (ЧЕРЕВНА ПОРОЖНИНА ТА нижні кінцівки).
Пульс на нижніх кінцівках слабкий, недостатнього наповнення або відсутній. Біль у нижніх кінцівках. Такий перерозподіл крові призводить до посиленого розвитку плечового пояса людини (широкі плечі, добре розвинені м’язи) з недостатнім розвитком нижньої частини, зокрема нижніх кінцівок (вони слабкі, м’язи їх погано розвинуті).
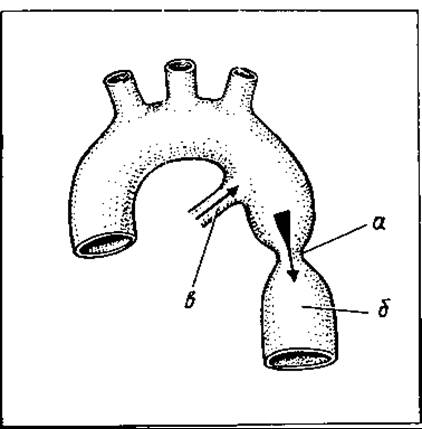
Мал. 135. Коарктація аорти: а — артеріальна протока; б — коарктація аорти; в — низхідна аорта
Хворі скаржаться на головний біль та оніміння в нижніх кінцівках і блідість їх шкіри. Фізикально вада виявляється різким систолічним шумом на аорті та гіпертрофією лівого шлуночка.
Багато хворих помирають від недостатності серця ще в ранньому віці, а в дорослому віці — від ускладнень гіпертензії.
За своєчасної діагностики вада успішно лікується хірургічним шляхом — резекцією звуженої частини аорти з прямим анастомозом (1—2 см звуження) або ж із застосуванням пластики. Виконуєшся операція без екстракорпорального кровообігу (без застосування апарата штучного кровообігу).
Рідкісною вадою розвитку аорти є подвоєння її дуги, яке спричинює компресію трахеї та стравоходу. Лікується хірургічно.
Дефект міжпередсердної перегородки — порівняно часта вада (5— 25 % від усіх вад). Він буває(переважно) високий, первинний, та низький, вторинний.
При такій ваді спостерігається викид крові з лівого в праве передсердя (внаслідок більшої еластичності правого шлуночка та легеневої артеріальної системи).
Малі за розмірами первинні дефекти можуть довго існувати без клінічних виявів, однак за більш-менш значних первинних та всіх вторинних дефектів уже в молодому віці розвивається серцева недостатність унаслідок посиленого легеневого кровоплину і перевантаження правого шлуночка. Хворі задихаються. Лікування їх полягає в хірургічному закритті дефекту на відкритому серці при штучному кровообігу.
Дефект міжшлуночкової перегородки зустрічається дещо частіше від дефекту міжпередсердної перегородки і локалізується або в мембранозній, або в м’язовій частинах перегородки. Він широко варіює від невеликого до повної відсутності перегородки (трикамерне серце). Як і при дефекті міжпередсердної перегородки, при цій ваді Кров надходить зліва направо — з лівого шлуночка, де тиск крові високий, в правий, в якому тиск крові нижчий. Це призводить до гіпертензії в легеневій артерії з подальшим розвитком недостатності правого шлуночка через перевантаження його.
У хворих вислуховується систолічний шум в третьому-четвертому міжребер’ї зліва. Серце збільшене. Супроводжується задишкою, особливо під час фізичного навантаження, серцебиттям та швидкою втомою. Лише у разі невеликих ізольованих дефектів не потрібна операція. Великі ж дефекти та комбіновані з іншими вадами серця потребують операції в ранньому віці (5—6 років), оскільки легенева Гіпертензія швидко прогресує і коли тиск досягає 60 мм рт.ст., операція стає протипоказаною, бо зашивання дефекту в такому разі зумовлює надмірне навантаження на правий шлуночок і раптовий його параліч.
Тетрада Фалло (вада описана французьким патологоанатомом Fallot у 1885 р.) є однією з найтяжчих багатокомпонентних вад «синього» чи «чорного» типу. Морфологічну основу його складають стеноз легеневої артерії, точніше інфуцдибулярний, тобто лійкоподібний, стеноз виходу з правого шлуночка, високий дефект міжшлуночкової перегородки, зміщення вправо (декстропозиція) аорти та гіпертрофія правого шлуночка. При цій ваді в легені внаслідок стенозу виходу з правого шлуночка потрапляє дуже мало крові, що зумовлює гіпоксемію. Крім того, через дефект у перегородці між шлуночками змішується венозна кров з артеріальною, що поглиблює гіпоксемію.
У крові накопичується багато редукованого гемоглобіну (понад 3 % замість 0,3 % в нормі), чим і спричинюється ціаноз. У дітей з цією вадою синюшний колір шкіри, спостерігаються прискорене серцебиття та дихання, РІСТ І РОЗВИТОК їх затримується, у них часто бувають так звані задишково-синюшні приступи, які можуть закінчуватись смертю. В позі навколішки хворим легше дихати.
Вислуховується грубий систолічний шум у третьому-четвертому міжребер’ї зліва від груднини, Грудна клітка деформована, пальці мають вигляд барабанних паличок. На рентгенограмі серце має форму «дерев’яного чобітка» — запала зона легеневої артерії та піднята і заокруглена верхівка серця. Без хірургічного втручання діти помирають у підлітковому віці.
Проводять хірургічну корекцію дефекту, частіше двоетапну (перший етап — накладання анастомозу між легеневою артерією вище від ділянки звуження з однією з гілок аорти, переважно підключичною, а через 1— 2 роки ліквідують радикально усі елементи вади).
ПОДВІЙНІ ТА ЧИСЛЕННІ ВИРОДЛИВОСТІ
Подвійні або численні виродливості є наслідком вад розвитку в порожнині матки двох (або більше) плодів, які розвиваються з кількох плідних яєць (за нормального розвитку це двояйцеві близнюки) або з одного (за нормального розвитку народжуються однояйцеві близнюки однієї статі).
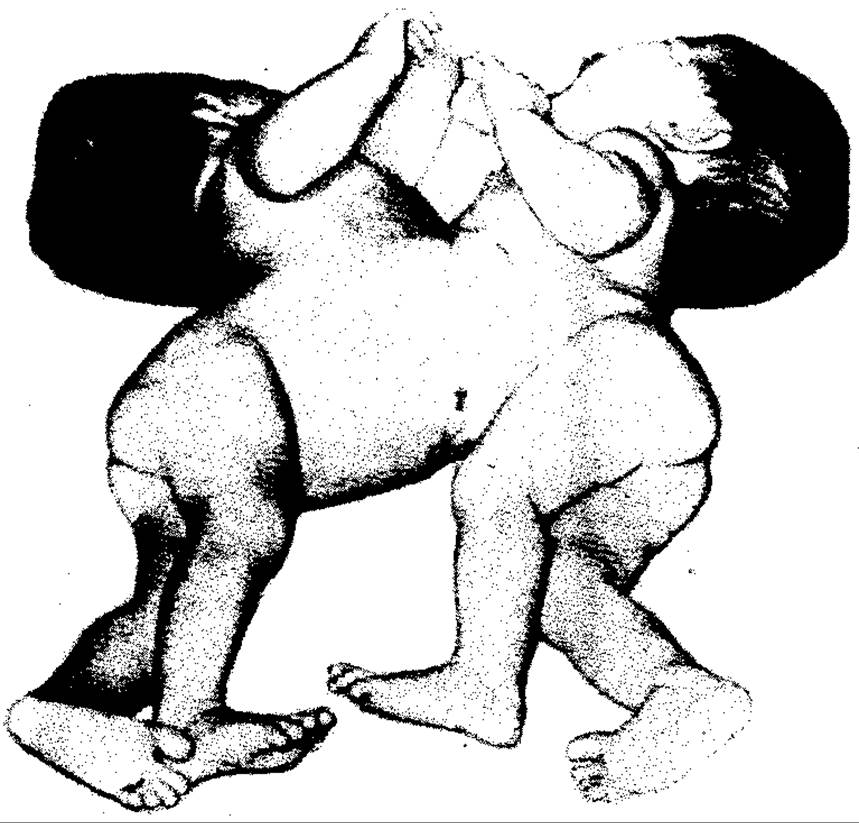
Мал. 136. Торакопаги (K.L.Mooze)
Подвійні виродливості можуть бути симетричними і несиметричними, вільними чи поєднаними. Серед асиметричних вільних плодів-близнюків один може бути нормально розвинений, другий недорозвинений. У недорозвиненого близнюка може не бути нижніх кінцівок або буває рудиментарне серце і голова, тому таких близнюків називають acardius, acephalus «без серця», «без мозку», або є тільки голова, тоді як тулуба і нижніх кінцівок немає (acardius acorms). Невільні симетричні подвійні виродливості являють собою однаковою мірою розвинуті організми, які з’єднані між собою різними частинами тіла. Часто у них спільні Кровообіг (серце), Головний мозок, Печінка, інші внутрішні органи чи частина тіла.
Деякі з цих організмів майже нормально сформовані і зрощені лише шкірою або кістками. Залежно від топографії зрощення розрізняють торакопаги (мал. 136; зрощені грудні клітки), стернопаги (зрощені грудниною), пігопаги (зрощені крижами), краніопаги (зрощені черепами) тощо. Це так звані паралельні подвоєння. Такі близнюки можуть жити дуже довго, прикладом чого є сіамські близнюки (стернопаги), що жили понад 60 років.
Бувають зрощення верхньої частини тіла з окремо сформованими лише нижньою частиною тулуба та кінцівками (цефалопаги та торакопаги) і, навпаки, відокремлені лише в ділянці голови два обличчя або дві голови (diprosopus та dicephalus) та зрощення нижнього кінця. Краніопаги зрощені лише головами.
Нещодавно канадські лікарі розділили таких близнюків.
Асиметричні подвійні виродливості становлять собою два нерівномірно розвинені організми: один майже нормальний, а другий недорозвинений, прикріплений до першого в різних ділянках тіла. Розвинений називається аутозитом, а недорозвинений — паразитом.
Залежно від ділянки прикріплення паразита вони мають назву: епігнатус (epignathus) — паразит, що прирощений до щелепи аутозита, pigopaguscrаnіо та thoracopagus — прирощені відповідно до крижів та промежини, черепа, грудей.
Іноді паразит перебуває в початковій стадії розвитку в середині нормального плода (плід у плоду). Коли внутрішній паразит являє собою лише безформну пухлину, яка має елементи різних тканин або органів, його називають тератомою, або організменою тератомою.
Лікування виродливостей, їх відокремлення звичайно хірургічне. У симетричних подвійних виродливостей воно можливе лише за наявності в кожного з них автономних органів життєзабезпечення.
Профілактика ПРИРОДЖЕНИХ ВАД РОЗВИТКУ
Запобігання вадам розвитку передбачає комплекс заходів як індивідуального, так і державного та глобального (планетарного) характеру.
Індивідуальні заходи, які легше піддаються регулюванню, полягають в здоровому способі життя та праці (нормальне харчування, викорінення шкідливих звичок), ефективному відпочинку, своєчасному проведенні медико- генетичного консультування.
Медико-генетичне консультування особливо потрібно тим, у чиїх сім’ях спостерігалось народження дітей з вадами розвитку.
Медико-генетичне консультування охоплює загальне лікарське обстеження (Анамнез, Огляд, дерматогліфіка - вивчення малюнків на шкірі долоні та пальців, генеалогічне, цитогенетичне, ультразвукове та ін.).
Сучасні дослідження, особливо ультразвукове та дослідження каріотипу плода шляхом амніотичної пункції, дозволяють діагностувати вади розвитку плода до народження. Виявлення вади дозволяє своєчасно інформувати про це сім’ю (матір) і залежно від характеру вади та ставлення до цього батьків перервати Вагітність або зберегти її.
Масові заходи із запобігання природженим вадам розвитку охоплюють широкий спектр державних і міждержавних акцій, серед яких найважливішими є оздоровлення екології (профілактика та усунення радіаційного забруднення повітря, води, землі; боротьба з забрудненням довкілля пестицидами, гербіцидами, токсичними відходами промислового виробництва тощо), суворе додержання правил запровадження в практику лікарняних засобів шляхом ретельного вивчення їх тератогенності, захист здоров’я працівників на шкідливих підприємствах, науково обгрунтовані та добре організовані умови праці та харчування людей, поліпшення медичної допомоги населенню, підвищення матеріального добробуту, культурного рівня та санітарної освіти народу тощо.
Последнее обновление: 14/02/2024
Редакционная и учебная адаптация: Данный материал сведен на основе первоисточника/оригинального текста. Команда проекта осуществила редакционную обзорную обработку, исправление технических неточностей, структурирование разделов и адаптацию содержания к учебному формату.
Что было обработано:
- устранение форматных дефектов (OCR-ошибки, разрывы структуры, дефектные символы);
- редакционное упорядочивание содержания;
- унификация терминов в соответствии с академическими источниками;
- проверка соответствия фактических утверждений текста первоисточнику.
Все упоминания об авторе, годе издания и происхождении первичного текста сохранены в соответствии с источником.