Основи медичної генетики - Бужієвська T.I. 2001
Спадкові хвороби
Первинні імунодефіцити
Клінічні прояви (фенотип) кожної, особливо інфекційної та алергічної патології, залежить від імуногенетичної програми індивідуума. Захисні імунні сили організму реалізуються в ході життєдіяльності 2∙1012 клітин, роботи 4-х специфічних органів (тимусу, кісткового мозку, селезінки та лімфатичних вузлів).
В-лімфоцити у відповідь на зустріч організму з антигеном синтезують специфічні антитіла, ІМУНОГЛОБУЛІНИ G, М,А, Е та D (залежно від будови важкого ланцюга молекули). Кожна молекула імуноглобуліну складається з чотирьох поліпептидних ланцюгів: 2 легких (має по одній варіабельній та константній частині) і 2 важких (по одній варіабельній та три константні частини). Гени, які кодують імуноглобуліни, утворюють З групи зчеплення (дві для легких ланцюгів та одна для важкого ланцюга) на різних хромосомах. Узгоджена експресія цих генів забезпечує специфічність та своєчасність Імунних реакцій організму. В молекулі імуноглобуліну варіабельні ділянки (V) двох ланцюгів (легкого —L, важкого—Н) створюють «пастки для антигенів», акцепторні закінчення. Константні (С) ділянки двох важких (Н) ланцюгів утворюють ефекторний кінець молекули, від якого залежить частка специфічного комплексу антиген + антитіло: аглютинація, лізис, зв'язування комплементу та ін.
Схематичне зображення молекули гаммаглобуліну подано на рис. 12.
Т-клітини залежно від функції, яку вони виконують, можуть бути помічниками (Т-хелпери), пригноблювачами (Т-супресори), вбивцями (Т-кілери). У цих клітинах працюють генимодифікатори імунної відповіді, що кодують інтерферони, інтерлейкіни та інші імуномодулятори. Мутації в різних генах різних клітин зумовлюють весь спектр первинних імунодефіцитів. Під час кожної імунної відповіді створюється клон спеціалізованих лімфоцитів, які зберігають пам'ять про антиген. Щоразу під час створення імунітету клітини повинні інтенсивно розмножуватися, тому повинна багаторазово реплікуватися ДНК (проходити її синтез). Дефект одного з ферментів, що бере участь в синтезі ДНК — аденозиндезамінази (забезпечує гідролітичне дезамінування аденозину) є причиною тяжкої комбінованої імунної недостатності (ТКІН).
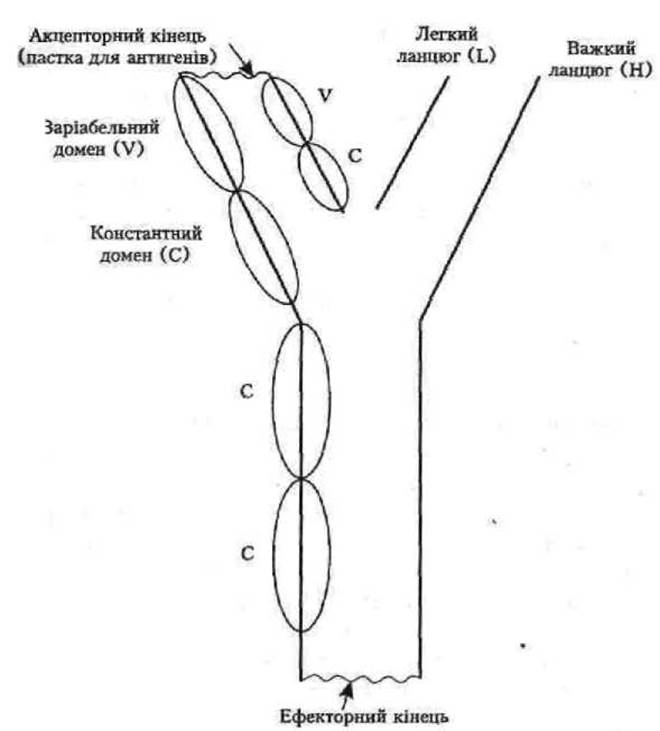
Рис. 12. Схема молекули гаммаглобулілу
ТКШ проявляється від народження дитини лімфопенією й аплазією вилочкової залози, відсутністю гіперчутливості уповільненого типу, порушенням синтезу імуноглобулінів. Лімфоцити хворих не реагують на мітогени in vitro. АР-тип успадкування. Етіологічне значення мають мутації в генах аденозиндезамінази та в гені активації рекомбінації (ГАР-1). Єдиним ефективним методом Лікування в наш час вважається трансплантація гістосумісного кісткового мозку. До операції хворі діти потребують забезпечення стерильних умов існування (стерильні їжа, повітря, скафандр і т. ін.).
Серед найбільш розповсюджених спадкових імунодефіцитів слід зупинитися на таких.
Агаммаглобулінемія, гіпогаммаглобулінемія — проявляються у віці від б місяців до 2 років хронічними (пневмонії, синусити) інфекціями, що викликаються незвичними, іноді навіть непатогенними збудниками. При цьому спостерігається дефіцит імуноглобулінів G. Можливі поствакцинальні ускладнення. Існують форми з АР- та ХР- (хвороба Брутона) типом успадкування.
Селективний дефіцит імуноглобулінів А — звичайно починає проявлятися в 3 —7-річному віці. З цим імунодефіцитним станом пов'язані: ревматоїдний артрит, червоний вовчак, анкілозивний спондиліт, дерматоміозит, атонічний дерматит, ячміні, отит, лямбліоз, Цукровий діабет, тиреоїдит та інші аутоімунні процеси. АР-тип успадкування, частина цих станів зчеплена з аномалією 18-ї Хромосоми.
Зміна співвідношення імуноглобулінів М та G вбік збільшення концентрації IgM характерна для лімфаденопатії.
При генетично зумовленому Ig Е-синдромі спостерігається непогана стійкість до звичайних інфекцій. Водночас у дітей поволі, майже непомітно з'являються холодні абсцеси, без болю та підвищення температури тіла, сверблячий дерматит, алергічні стани, кандидоз.
У дітей до 2 — 3 міс життя може спостерігатися транзиторна гіпогаммаглобулінемія, яка пов'язана з пізнім імунологічним стартом.
Імунопроліферативна хвороба — проявляється чутливістю до вірусу Епстейна-Барра та схильністю до захворювання на лімфому (навіть злоякісну) та інфекційний мононуклеоз. ХР-тип успадкування.
Гіпоплазія тимуса (синдром Ді Джорджі). З одного боку — це ПВР, з іншого — Т-клітинний імунодефіцит. Проявляється з народження судомами (гіпопара- тиреоз), підвищеною схильністю до вірусних та грибкових інфекцій, аномаліями розвитку кісткової системи, ротової порожнини, внутрішніх органів, аплазією або гіпоплазією вилочкової залози, недостатністю Т-клітинного імунітету. Без відповідного лікування смерть хворого настає до 2 років життя. Описані родини з АР- або АД-типом успадкування, делецією хромосоми 22q 11, спостерігаються спорадичні випадки.
Синдром Віскотта—Олдріча — імунодефіцит з тромбоцитопенією та екземою. Клінічно проявляється з народження: тромбоцитопенічна пурпура, рідкі випорожнення з домішками крові, згодом приєднуються Пневмонія, отит, дерматити, екзема, можливе виникнення злоякісних пухлин, що нагадує ретикулоендотеліоз. Провідним є геморагічний синдром, генетичний'блок на рівні В-лімфоцитів. Тип успадкування — ХР.
Атаксія-телеангіектазія (синдром Луї—Бара) — комплексне захворювання імунної, нервової та ендокринної систем з частим ушкодженням шкіри та печінки. Одним з симптомів цієї патології є резистентний до інсуліну цукровий діабет, схильність до злоякісних пухлин ретикулярного типу. АР-тип успадкування.
Лікування імунодефіцитів, головним чином, симптоматичне: вітамінотерапія, антибіотики, гемотрансфузія; трансплантація кісткового мозку в наших умовах проблематична; імуномоделююча терапія може і буде використовуватися все більше в зв'язку з досягненнями генно-інженерної промисловості та зростаючими можливостями діагностики імунного статусу (дзеркала) кожного хворого.
Последнее обновление: 05/02/2024
Редакционная и учебная адаптация: Данный материал сведен на основе первоисточника/оригинального текста. Команда проекта осуществила редакционную обзорную обработку, исправление технических неточностей, структурирование разделов и адаптацию содержания к учебному формату.
Что было обработано:
- устранение форматных дефектов (OCR-ошибки, разрывы структуры, дефектные символы);
- редакционное упорядочивание содержания;
- унификация терминов в соответствии с академическими источниками;
- проверка соответствия фактических утверждений текста первоисточнику.
Все упоминания об авторе, годе издания и происхождении первичного текста сохранены в соответствии с источником.