МИКРОБИОЛОГИЯ БИОЛОГИЯ ПРОКАРИОТОВ ТОМ III - А. В. ПИНЕВИЧ - 2009
ГЛАВА 17. ОНТОГЕНЕЗ
17.2. Старение и смерть клетки
Прокариотов считают потенциально бессмертными — из-за отсутствия у них комплекса феноменов старения и смерти, свойственного высшим организмам. Так ли это на самом деле, и не существуют ли у прокариотов аналоги процессов старения и смерти?
17.2.1. Старение клетки
В стационарной фазе роста (см. раздел 21.1.1) бактерии перестают размножаться — чаще всего, по причине истощения питательных субстратов. Такие «стерильные» клетки постепенно теряют жизнеспособность. Данное физиологическое состояние называется условным, старением из-за прекращения роста (англ. conditional senescence elicited by growt.h arrest).
Рост и выживание (англ. maintenance) являются двумя взаимно конкурирующими физиологическими состояниями. Внутренним сигналом тревоги, или алармоном
(англ. alarmone, от alarm — тревога), который прекращает рост и ориентирует клетку на выживание, служит (p)ppGpp (см. рис. 254).
Регуляторное влияние (p)ppGpp на транскрипцию объясняется аллостерическим связыванием этого нуклеотида с кор-ферментом РНК-полимеразы (рис. 225).
Рис. 225. Выбор между ростом и выживанием, зависящий от уровня (р)ррGрр. Е — кор-фермент РНК-полимеразы; RelА и SроТ — соответственно, синтетазы I и II.
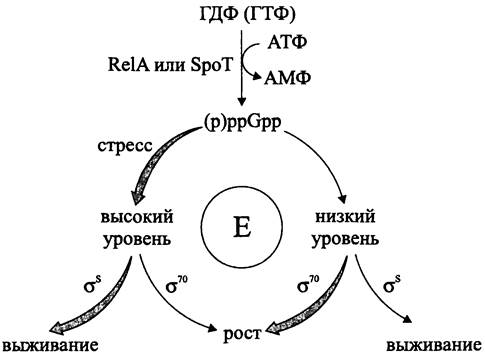
У бактерий, в том числе Е. coli, содержание (p)ppGpp может регулироваться разными способами; в частности, оно повышается при строгом ответе на аминокислотное голодание (см. раздел 18.3).
Реакцию ГДФ (ГТФ) + АТФ —> (p)ppGpp + АМФ катализирует синтетаза I (RelA). Активатором этого фермента служат «незаряженные» тРНК, которые накапливаются при аминокислотном голодании или в результате ингибирования синтеза отдельных аминокислот. Содержание (p)ppGpp возрастает и при углеродном голодании, однако в данном случае используется не синтетаза I, а синтетаза II (SpoT).
В основе выбора между ростом и выживанием лежит конкуренция главного транскрипционного фактора σ70 и альтернативного транскрипционного фактора σ38 (σs) за связь с кор-ферментом РНК-полимеразы (см. раздел 16.7.1). Голофермент, содержащий σ70, связывается с промоторными участками «обычных» генов домашнего хозяйства, продукты транскрипции которых обслуживают ростовые процессы. В свою очередь, голофермент, содержащий σs, связывается с промоторными участками «стрессовых» генов. Избыточный синтез σ70 способствует росту, но в то же время делает клетку чувствительной к стрессорам. При низком содержании (p)ppGpp в состав голофермента преимущественно входит σ70, а при высоком содержании — σs.
Таким образом, (p)ppGpp обеспечивает соответствие между активностью РНК- полимеразы и трофическими ресурсами окружающей среды, что и определяет выбор между ростом и выживанием (рис. 225). Когда трофический статус благоприятен (низкий уровень (p)ppGpp), с помощью σ70 транскрибируются гены домашнего хозяйства, и клетки растут. В свою очередь, при неблагоприятном трофическом статусе (высокий уровень (p)ppGpp) с помощью σS транскрибируются стрессовые гены, и клетки от роста переходят к выживанию.
Старение клеток при выживании в стационарной фазе связано с потерей жизнеспособности из-за снижения интенсивности метаболических процессов и нарушения целостности СМ. В настоящее время основной физиологической причиной старения считается окислительный стресс из-за накопления активных форм кислорода (см. раздел 19.1.6). На молекулярном уровне старение связано не только с переходом полноценных белков в окисленную форму, но и с нарушением их фолдинга (см. I том учебника).
Интересно, что патологическая смерть бактериальной клетки под воздействием бактерицидных антибиотиков (например, пенициллина, ингибирующего синтез муреина) осуществляется с помощью такого же генерального механизма, как и при старении, и связана с окислительным стрессом, при котором накапливаются активные формы кислорода. В то же время при воздействии бактериостатических антибиотиков (например, рифампицина, ингибирующего транскрипцию) активные формы кислорода не накапливаются.
17.2.2. Смерть клетки
Жизненные циклы бактерий в ряде случаев, в том числе при старении, завершаются запрограммированной смертью, которая может принимать форму апоптоза.
Запрограммированная смерть (англ. programmed cell death, PCD) и ее характерное фенотипическое выражение, апоптоз (греч. apoptosis — устранение) являются ничем иным, как самоубийством. Это общебиологический феномен, при котором, клетки, по той или иной причине оказавшиеся дефектными, элиминируются из популяции.
Слова «запрограммированная смерть клетки» и «апоптоз» часто используются в качестве синонимов. Однако на самом деле понятие «запрограммированная смерть» охватывает более широкий круг биологических явлений. К ним, в частности, относится и апоптоз, который представляет собой комплекс морфологических изменений, характерных для клетки, умирающей по эндогенной программе.
Термин «апоптоз», предложенный в 1972 г. английским физиологом Керром (J. F. R. Kerr), традиционно используется применительно к запрограммированной клеточной смерти у ядерных, особенно многоклеточных организмов. К числу морфо-биохимических признаков апоптоза относятся разбухание клетки, конденсация хроматина, протеолиз и деградация ДНК. Диагностическим признаком апоптоза служит процессинг ядерной ДНК с образованием «лестницы» фрагментов в размерном диапазоне 180-200 п. н.
Апоптоз соответствует физиологической смерти к легки, в отличие от некроза (греч. necrosis — смерть), или патологической смерти, вызванной внешним повреждающим воздействием. При апоптозе мембраны, в частности мембрана лизосомы, остаются интактными, и завершающей фазой служит распад клетки с образованием апоптозных телец, которые поглощаются фагоцитами; содержимое мертвой клетки не выходит наружу, что исключает развитие воспаления. При некрозе, напротив, лизосомы разрушаются; содержимое клетки выходит наружу, вызывая воспаление. Таким образом, в отличие от апоптоза, некроз повреждает соседние клетки.
Апоптоз — это альтруистический процесс, позволяющий устранять ненужные клетки ради эффективного функционирования или выживания многоклеточного организма. Он неразрывно связан с дифференцировкой, гистогенезом и органогенезом, а также уничтожает опасные клетки (мутантные, неопластические, инфицированные вирусами и т. д.). Индукторами апоптоза служат как внутриклеточные, так и внеклеточные сигналы, в частности цитокины (интерфероны, интерлейкины, факторы некроза опухоли и др.), а также гормоны. «Сигналы о смерти» поступают в митохондрии, что приводит к коллапсу протонного градиента и высвобождению белковых факторов проапоптоза, в частности растворимого цитохрома с. Центральную роль в механизме апоптоза играют цистеиновые киллер-протеазы, принадлежащие к семейству каспаз (англ. caspase; от cysteinyl aspartate-specific proteinase).
В случае одноклеточных организмов, особенно прокариотов, об апоптозе говорят редко по причине отсутствия у них соответствующего комплекса морфо-биохимических признаков клеточной смерти или несходства этих признаков с эукариотными признаками апоптоза (хотя у прокариотов также наблюдаются дисперсия нуклеоида, деградация внутриклеточных структур, плазмолиз и фрагментация СМ). В данном случае применяется только термин «запрограммированная смерть клетки», которым в настоящее время обозначают любую форму клеточной смерти по эндогенной программе, независимо от того, чем она запускается, а также независимо от того, имеет ли картина смерти признаки классического апоптоза.
Запрограммированная смерть у прокариотов регулируется генетически и связана с синтезом или активацией ферментов, участвующих в цепи превращений, ведущих к гибели клетки. Конкретный характер этих превращений зависит от специфики онтогенеза, метаболического статуса и присутствия сателлитных геномов.
Некоторые из систем, вызывающих запрограммированную смерть прокариотной клетки, широко распространены, тогда как другие уникальны. Как правило, они представляют собой белковый токсин, действующий в паре с антитоксином (белком или антисмысловой РНК). Механизм действия токсина может быть разнообразным — от рестрикции ДНК до разрушения клеточной оболочки.
Запрограммированной смерти, в частности, подвергаются:
— вегетативные клетки миксобактерий при формировании плодового тела;
— клетки субстратного мицелия актинобактерий при формировании воздушного мицелия;
— клетка-спорангий при дифференциации эндоспоры;
— сегрегант, излеченный от плазмиды;
— вегетативные клетки в стационарной фазе;
— клетка-хозяин при репродукции бактериофага.
Смерть вегетативных клеток миксобактерий при формировании плодового тела. В случае миксобактерий, а также актинобактерий (см. ниже) альтруистический автолиз отдельных клеток обеспечивает остальную популяцию питательными субстратами, давая возможность завершить клеточный цикл образованием дифференцированных особей.
Выше мы уже говорили о том, что миксобакгерии обладают сложным циклом развития. У наиболее изученных видов, Myxococcus xanthus и Stigmatella aurantiaca, скопление клеток погружено в слизистый матрикс, и такая «улитка» медленно ползет по увлажненному твердому субстрату (см. I том учебника). В метаболическом отношении миксобактерий являются либо сапрофитами, переваривающими мертвые тела других бактерий с помощью комплекса внеклеточных гидролаз, либо хищниками, которые лизируют живых бактерий и питаются содержимым их клеток. При истощении питательных субстратов (сигналом об этом служит повышенный уровень (p)ppGpp; см. выше) миксобактерий начинают взаимно агрегировать с образованием спорангиев, наполненных округлыми или эллипсоидными цистами, или миксоспорами — продуктами дифференциации палочковидных вегетативных клеток (см. раздел 17.5.2.1).
Ценой образования миксоспор является смерть значительной части, иногда до 90%, вегетативных клеток из-за секреции автоцида (англ. autocide) — смеси разветвленных жирных кислот, представляющих собой побочный продукт обновления мембранных липидов. Такое альтруистическое самоубийство в сочетании с агрегацией обеспечивает остальную часть популяции концентрированными питательными субстратами, что позволяет завершить дифференциацию образованием спорангиев, плодовых тел и миксоспор.
Смерть клеток субстратного мицелия у актинобактерий при формировании воздушного мицелия. Высшие актинобактерии (см. I том учебника) проявляют сходство с миксобактериями, образуя окрашенные макроколонии на поверхности твердых субстратов. В начальной фазе развития они образуют субстратный мицелий (англ. substrate mycelium), состоящий из (не)ветвящихся гиф с множественными нуклеоидами и редко расположенными септами.
При дефиците питательных субстратов (сигналом об этом, как обычно, служит повышенный уровень (p)ppGpp) актинобактерии прекращают расти, что сопровождается альтруистическим автолизом части гиф. Переход к образованию воздушного мицелия (англ. arial mycelium) связан с чувством кворума, наступающим при накоплении феромонов (см. раздел 18.4.2). Гифы воздушного мицелия обладают повышенным тургором благодаря деполимеризации резервных полиглюкозидов, а также имеют липофильную поверхность, что связано с образованием специализированных белков, ассоциированных со спорами (англ. spore-associated protein, SAP). Это позволяет гифе расти вверх, преодолевая поверхностное натяжение водной пленки (см. раздел 17.3.2).
Росту воздушного мицелия в экстремальной среде предшествуют гидролиз биомассы мертвых гиф и поглощение образующихся субстратов сохранившейся частью гиф (как и в случае миксобактерий, такое поведение эквивалентно каннибализму). Поскольку гифы воздушного мицелия не могут поглощать из воздуха питательные субстраты, они прекращают рост и переходят к дифференцации. Цитологической основой этого процесса является апикальное септирование. Нуклеоиды поодиночке изолируются в коротких отрезках воздушных гиф, которые дифференцируются в пигментированные цисты, или так называемые споры (см. ниже). После автолиза нефрагментированных участков воздушных гиф споры освобождаются, а затем пассивно или активно, за счет плавания, распространяются в окружающей среде.
Смерть клетки-спорангия при дифференциации эндоспоры. Спорогенез В. subtilis обеспечивается ~60 генами, которые неупорядоченно распределены по хромосоме. Чаще всего триггером служит снижение концентрации питательных субстратов при переходе к стационарной фазе роста.
Процесс спорообразования подразделяется на обратимую фазу (стадии 0-II) и необратимую фазу (стадии III-VII), причем «точка возврата», при условии снабжения материнской клетки питательными субстратами, предшествует асимметричному делению. Ключевым моментом в спорообразовании служит переход регуляторного белка Spo0A в фосфорилированную форму, которая активирует транскрипцию генов спорообразования или подавляет транскрипцию ингибиторов спорообразования AbrB и Нрг (см. раздел 17.5.2.2).
В клетке-спорангии синтезируются специфические белки, в частности ключевой альтернативный фактор транскрипции σк. Последующую взаимную координацию дифференциации спорангия и преспоры обеспечивает взаимодействующая система сигма-факторов (см. раздел 17.5.2.2). Установлено, что причиной смерти спорангия становятся автолизины, в частности амидаза CwlC (сокр. англ. cell wall lysis), хотя механизм, запускающий автолиз, еще не выяснен.
Смерть сегреганта, излеченного от плазмиды: смертельные/антисмертельные факторы плазмидной природы. Генетические системы такого типа содержатся в низкокопийных плазмидах; они обнаружены только у Е. coli. Во всех случаях они состоят из двух генов, кодирующих, соответственно, стабильный токсин и нестабильный антитоксин (антидот).
Плазмидные гены, которые кодируют токсин/антитоксин, получили образное название — модули привыкания (англ. addiction module), поскольку клетка, как наркоман, зависит от постоянного присутствия этого необязательного генетического элемента.
Когда модуль привыкания имеет плазмидную локализацию, сегреганты, по той или иной причине не получившие плазмид(ы), погибают (англ. postsegregational killing effect). Летальный эффект объясняется тем, что противоядие разрушается быстрее, чем токсин. Логика этого явления очевидна — механизм привыкания косвенно обеспечивает стабильное наследование плазмид, наряду с такими механизмами прямого действия, как системы контроля репликации, системы расхождения сестринских плазмид и системы декатенации (см. разделы 16.4.1.2 и 16.4.2.2). Эгоистичное поведение плазмид гарантирует их сохранение, поскольку в отсутствии программируемого самоубийства быстрее размножающиеся «излеченные» клоны вытеснили бы клоны с плазмидами.
Модули привыкания бывают двух типов. В первом случае оба продукта экспрессии, стабильный токсин и нестабильный антитоксин, являются белками. Такие модули привыкания называются «белковыми» системами киллерных генов (англ. protec killer gene System). Во втором случае токсин также является стабильным белком, однако антитоксин представляет собой нестабильную антисмысловую РНК из семейства hok/sok (сокр. англ. host-killing/supressor of killing), которая препятствует трансляции мРНК токсина в клетках, содержащих плазмиды.
«Белковые» системы киллерных генов имеют сходную генетическую структуру, взаимно гомологичны (что говорит об их дивергентном, а не конвергентном происхождении) и имеют ряд общих свойств:
— продуктом upstream-гена является нестабильный антитоксин, а продуктом downstream-гена стабильный токсин;
— токсин и антитоксин представляют собой небольшие белки, соответственно, из 100-130 и 70 -85 а. о.;
— антитоксин синтезируется в избыточном количестве и разрушается специфической протеазой;
— токсин и антитоксин непосредственно взаимодействуют друг с другом;
— модуль привыкания авторегулируется на уровне транскрипции комплексом токсин/антитоксин или только антитоксином.
Специфика отдельных систем киллерных генов связана со свойствами токсина/антитоксина, типом разрушающей антитоксин протеазы и клеточной мишенью для токсина.
Наиболее изученной белковой системой киллерных генов является модуль привыкания ccdAB (сокр. англ. control of cell death). Он содержится в однокогшйной конъюгативной F-плазмиде размером 95 т. п. н. (см. разделы 16.1.2.1 и 16.6.1.3). Гены ccdA и ccdB кодируют, соответственно, антитоксин CcdA (8,7 кДа) и токсин CcdB (11,7 кДа). Помимо оиерона-модуля ccdAB, F-плазмида содержит два вспомогательных оперона — smB (сокр. англ. stabile RNA dégradation) и flm (сокр. англ. F leading maintenance). Эти опероны представляют собой модули привыкания, где роль антидота выполняет антисмысловая РНК. Эффект убийства выражается в гибели >90% бесплазмидных сегрегантов. В отсутствии антитоксина CcdA токсин CcdB вызывает образование нитевидных клеток и индуцирует SOS-ответ, что приводит к апоптозу. Непосредственной мишенью для токсина служит каталитическая субъединица ДНК-гиразы GyrA (топоизомеразы типа-II), которая обеспечивает АТФ-зависимую суперспирализацию хромосомы, делая двухцепочечный разрез ДНК, пропуская сквозь него другой участок и сращивая тупые концы (см. разделы 16.4-1.1 и 16.4.2). Результатом связывания токсина с гиразой GyrA становится летальная релаксация суперспирализованной ДНК. Антитоксин, который синтезируется в «плазмидных» клетках, предотвращает эффект токсина, образуя с ним прочный комплекс. Авторегуляция транскрипции генов ccdАВ происходит за счет взаимодействия этого комплекса с промоторным сайтом. Деградацию антитоксина CcdA осуществляет АТФ-зависимая протеаза Lon (сокр. англ. lambda omission of N; название связано с тем, что субстратом этого фермента служит один из нескольких быстро деградирующих белков, в частности антитерминатор N фага лямбда).
Попутно отметим, что протеаза Lon принадлежит к суперсемейству ААА-АТФаз (см. раздел 16.6.1.3) и представлена во всех трех доменах глобального древа. Она разрушает широкий круг нестабильных белков, обеспечивающих, в числе прочего, организацию, воспроизведение и экспрессию генетических структур.
Аналогичный механизм собственного стабильного сохранения с помощью белковой системы киллерных генов используют низкокопийные плазмиды R1 и R100, принадлежащие к группе несовместимости IncFII (см. раздел 16.1.2.1). В плазмиде RI модуль привыкания kis/kid (сокр. англ. killing suppression/killing déterminant) кодирует токсин Kis (12 кДа) и антитоксин Kid (9,3 кДа). Сходным образом, в плазмиде R100 модуль привыкания pemlK (сокр. англ. plasmid emergency maintenance inhibitor/'plasmid emergency maintenance killing) кодирует токсин PemK (12 кДа) и антитоксин PemI (9,3 кДа). Для лучше изученного модуля привыкания pemlK установлено, что мишенью токсина PemK служит ДНК-геликаза DnaB, которая превращает открытый комплекс в препраймерный, расплетая двойную спираль ДНК (см. раздел 16.4.1.1). Таким образом, токсин выступает в роли ингибитора репликации, что вызывает летальный эффект, в то время как антитоксин нейтрализует его действие. Показано, что транскрипция генов pemlK негативно авторегулируется комплексом белков PemlK за счет блокирования промоторного сайга. Установлено также, что антитоксин PemI разрушается АТФ-зависимой протеазой Lon.
Похожий пример связан со стабильно наследуемой 4-7 копийной плазмидой РК2 (RP4). Ее модуль привыкания parDE кодирует токсин РагЕ (12 кДа) и антитоксин ParD (9 кДа). В отличие от предыдущих примеров, мишень для токсина не установлена, а антитоксин разрушается протеазой, не относящейся к семейству Lon.
Смерть вегетативных клеток в стационарной фазе. «Белковые» системы киллерных генов могут иметь не только плазмидную, но и хромосомную локализацию; в данном случае это гены самоубийства. Мы уже говорили о том, что при аминокислотном голодании содержание сигнального медиатора (p)ppGpp возрастает в результате активации синтетазы RelA. В хромосоме Е. сoli ген relA входит в общий оперон с генами mazEF (сокр. евр. ma-ze — что такое; функция этих генов вначале была неизвестна). Другое обозначение этих генов — chpAB (сокр. англ. chromosome homologue of реm; продукты их экспрессии гомологичны продуктам экспрессии генов модуля привыкания pemlK).
Ген mazF кодирует стабильный токсин (12,1 кДа), а ген mazE — нестабильный антитоксин (9,4 кДа), который разрушается под воздействием сериновой протеазы С1рР. Мишень для действия токсина MazF еще не установлена. Транскрипция генов mazEF негативно авторегулируется комплексом белков MazEF за счет блокирования промоторного сайта.
При азотном или углеродном голодании повышается уровень (p)ppGpp (см. рис. 225). Этот алармон связывается с кор-ферментом РНК-полимеразы и ингибирует экспрессию генов mazEF. Поскольку антитоксин MazE более лабилен, чем токсин MazF, преимущественное накопление последнего приводит к клеточной смерти. Иными словами, mazF — это регулируемый модуль привыкания.
Смерть хозяйской клетки при репродукции бактериофага. В конце цикла репродукции бактериофага происходит лизис хозяина. Этот биохимический процесс осуществляется с помощью двух ферментов, играющих роль токсинов, причем оба они кодируются геномом фага. Это холин (англ. holine; от hole — отверстие; не путать с холином-амином), который образует бреши в СМ, и эндолизин (англ. endolysine), который получает доступ к муреиновому саккулусу и вызывает его локальный гидролиз. Преждевременное действие этих ферментов, которое могло бы разрушить клетку до окончательного формирования фаговых частиц, предотвращается белками-антидотами. Как и другие антидоты, они лабильны, и к моменту завершения репродукции фага их концентрация опускается ниже критического уровня.
В то время как сборка и выход зрелых фаговых частиц обычно связаны со смертью хозяина, при лизогении (см. разделы 16.6.1.2 и 16.6.1.3) клетка остается интактной. Одним из немногих генов профага лямбда, активных в лизогенном состоянии, является ген гехВ (сокр. англ. rll exclusion; гех-оперон обеспечивает иммунитет к вторичному заражению фагом T4rII). Продукт экспрессии гена гехВ ингибирует расщепление двух ключевых субстратов протеазы СlрР — нестабильного белка ![]() , участвующего в репликации фаговой ДНК, и клеточного антитоксина MazE. Таким образом, эгоистичное поведение фага спасает хозяйскую клетку от смерти.
, участвующего в репликации фаговой ДНК, и клеточного антитоксина MazE. Таким образом, эгоистичное поведение фага спасает хозяйскую клетку от смерти.