Фізіологія людини - Вільям Ф. Ґанонґ 2002
Ендокринна система, метаболізм і репродуктивна функція
Ендокринні функції підшлункової залози й регулювання метаболізму вуглеводів
Наслідки нестачі інсуліну
Далекосяжні фізіологічні ефекти інсуліну пояснюють з огляду на передбачувані всебічні та важливі наслідки дефіциту інсуліну.
У людей дефіцит інсуліну є поширеним і серйозним патологічним станом. У тварин він може бути спричинений панкреатектомією; уведенням алоксану, стрептозоцйну та інших токсинів, що в певних дозах зумовлюють вибіркове руйнування В-клітин панкреатичних острівців; уведенням препаратів, що інгібують секрецію інсуліну; уведенням антиінсулінових антитіл. Відомі та описані деякі лінії мишей, щурів, хом’яків, морських свинок та мавп, що мають часту захворюваність на спонтанний цукровий діабет.
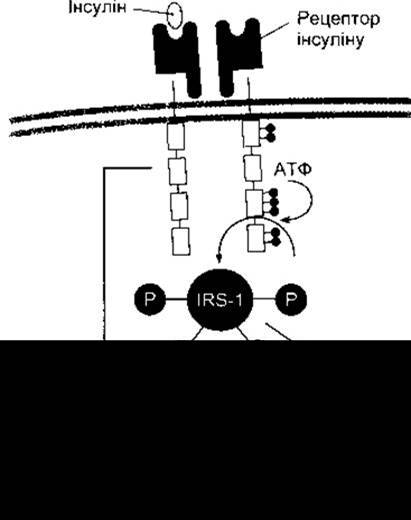
Рис. 19-7. Внутрішньоклітинні відповіді на зв’язування інсуліну з його рецептором. Темні кульки та кульки з буквою Р позначають фосфатні групи; IRS-1 - субстрат-1 інсулінового рецептора.
Сукупність аномалій, спричинених нестачею інсуліну, називають цукровим діабетом. Грецькі та римські лікарі використовували термін діабет для опису станів, головною ознакою яких був великий об’єм сечі. Розрізняли такі два типи: цукровий діабет, за якого сеча мала солодкий смак, та нецукровий діабет, за якого сеча не мала смаку. Тепер термін нецукровий діабет використовують для станів, за яких виникає дефіцит утворення або дії вазопресину (див. Розділ 14), а саме слово діабет є синонімом до цукрового діабету.
Діабет супроводжується поліурією, полідипсією, втратою маси за поліфагії (збільшеного апетиту), гіперглікемією, глюкозурією, кетонемією, ацидозом та комою. Є різноманітні біохімічні аномалії, проте головні дефекти, до яких можна звести всі аномалії, - це, по-перше, зменшене надходження глюкози до різних периферійних тканин та, по-друге, збільшене виділення печінкою глюкози в Кров’яне русло. Отже, виникає позаклітинний надлишок глюкози, а в багатьох клітинах - внутрішньоклітинна нестача глюкози, стан, який назвали голодуванням серед достатку. Також зменшується надходження амінокислот у м’язи та збільшується ліполіз.
Тепер відомо, що в разі діабету виникає абсолютна чи відносна гіперсекреція глюкагону. Це відбувається навіть за умов видалення підшлункової залози, оскільки Глюкагон, крім підшлункової залози секретується також у інших відділах травної системи. Соматостатин зменшує секрецію глюкагону та інсуліну (див. Розділ 14), і коли його вводять тваринам із видаленою підшлунковою залозою, то рівень глюкози в плазмі спадає до норми. Здається, що позаклітинний надлишок глюкози є частково наслідком гіперглюкагонемії. Проте деяка гіперглікемія простежується навіть тоді, коли секреція глюкагону зменшена до нуля.
Толерантність до глюкози
У разі діабету глюкоза накопичується в крові, особливо після їжі. Якщо діабетик піддається дії глюкозного навантаження, то його рівень глюкози у плазмі зростає й повертається до початкового рівня повільніше, ніж у нормальних людей. Відповідь на стандартну оральну тестову дозу глюкози - тест на толерантність до глюкози, - використовують у клінічному діагностуванні діабету (рис. 19-8).
Погіршена толерантність до глюкози у разі діабету частково є наслідком зменшеного надходження глюкози в клітини (зменшена периферійна утилізація). Без інсуліну надходження глюкози в скелетні, серцевий і гладкі м’язи та інші Тканини зменшується (рис. 19-9). Поглинання глюкози в печінці також зменшується, однак результат цього опосередкований. Абсорбція глюкози в кишці не змінюється, як і реабсорбція із сечі клітинами проксимальних канальців нирок. Поглинання глюкози більшою частиною мозку та еритроцитами також у нормі.
Другою, і головною, причиною гіперглікемії в разі діабету є розлад глюкостатичної функції печінки (див. Розділ 17). Печінка поглинає глюкозу з крові і запасає її у вигляді глікогену, та оскільки вона містить глюкозо-6-фосфат, то також вивільняє глюкозу в кров. Справді, Клод Бернард говорив про печінку як про ендокринну залозу, що секретує глюкозу. Інсулін сприяє синтезуванню глікогену і перешкоджає печінковому вивільненню глюкози. Якщо плазма вміщує багато глюкози, то зазвичай збільшується секреція інсуліну, і печінковий глюкогенез зменшується. Цей ефект зникає внаслідок діабету. Глюкагон теж робить внесок у гіперглікемію; якщо стрес чи хвороба серйозні, то печінкове вивільнення глюкози стимулюють катехоламіни, кортизол та Гормон росту.
Ефекти гіперглікемії
Сама по собі гіперглікемія може зумовити симптоми внаслідок гіперосмолярності крові. Крім того, якщо ниркова здатність реабсорбції глюкози зменшується, то виникає глюкозурія. Екскреція осмотично активних молекул глюкози супроводжується втратою великих кількостей води (осмотичний діурез; див. Розділ 38). Спричинена дегідратація активує механізми регулювання споживань води, що веде до полідипсії. Простежується помітна втрата Na+ та К+ із сечею. На кожний грам екскретованої глюкози Організм втрачає 4,1 ккал енергії. Збільшене споживання їжі для покриття цих втрат просто підвищує рівень глюкози в плазмі з наступним посиленням глюкозурії, не перешкоджаючи мобілізації ендогенних білків та жирових нагромаджень, а також втраті маси.
Якщо плазматичний рівень глюкози епізодично підвищується, то невеликі кількості гемоглобіну А неензимно глікозилюють з утворенням НbАІС (див. Розділ 27). Старанний контроль діабету за допомогою інсуліну знижує цей рівень, тому концентрацію НbАІС у клініці використовують як інтегрований показник діабетичного контролювання для періоду тривалістю чотири-шість тижнів до вимірювань.
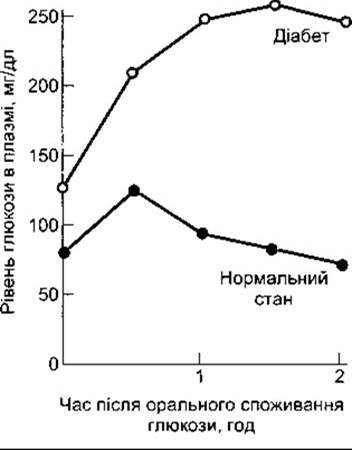
Рис. 19-8. Оральний тест на глюкозну толерантність. Дорослим дають 75 г глюкози на 300 мл води. В нормальних осіб венозний плазматичний рівень глюкози натще становить менше 115 мг/дл, через дві години після Введення - менше 140 мг/дл, і жодне із значень не перевищує 200 мг/дл. Цукровий діабет є, якщо двогодинне і будь-яке інше значення перевищує 200 мг/дл. Зменшена толерантність до глюкози виникає тоді, коли значення є більшими, ніж верхня межа норми, однак меншими від діагностичних значень діабету.
Значення хронічної гіперглікемії у формуванні тривалих ускладнень діабету розглянуто нижче.
Ефекти внутрішньоклітинного дефіциту глюкози
Надлишок глюкози поза клітиною в разі діабету контрастує з її внутрішньоклітинною нестачею. Катаболізм глюкози, зазвичай, є головним джерелом енергії для клітинних процесів і у випадку діабету енергетичні потреби можуть бути задоволені тільки завдяки використанню запасів білка й жиру. В цьому разі відбувається активування механізмів, що сильно підвищують катаболізм білків і жирів, один із наслідків підвищеного катаболізму жирів - кетонемія.
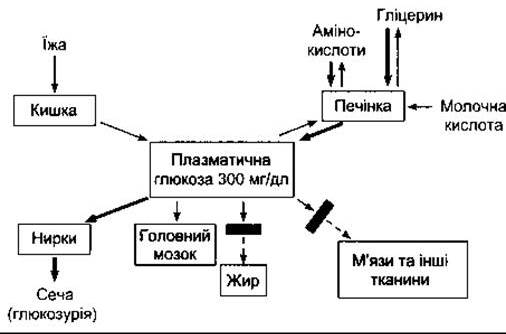
Рис. 19-9. Розлади гомеостазу плазматичної глюкози у разі дефіциту інсуліну. Порівняйте з рис. 17-14. Грубі стрілки показують реакції, описані в тексті. Прямокутники поперек стрілок позначають блоковані реакції.
Дефіцит утилізації глюкози в клітинах гіпоталамних вентромедіальних ядер є, можливо, причиною гіперфагії в разі діабету. Якщо активність центру насичення цього ядра послаблюється внаслідок зменшення утилізації глюкози в клітинах, то латеральна зона апетиту діє, не зазнаючи опору, і споживання їжі збільшується (див. Розділ 14). Проте є й інші пояснення цього явища.
Виснаження глікогену є звичним наслідком дефіциту глюкози. Звісно, що вміст глікогену в печінці та скелетних м’язах у діабетичних тварин знижений.
Зміни в метаболізмі білків
У випадку діабету швидкість, із якою відбувається ката- болізування амінокислот до СO2 і Н2O, зростає. Окрім цього, більше амінокислот перетворюється в глюкозу у печінці. Ці зміни показані на рис. 19-10, де також відображено інші головні аномалії проміжного обміну в печінці.
Деякі значення швидкості глюконеогенезу в діабетичної тварини, що голодує, можна отримати вимірюванням співвідношення кількості глюкози (декстрози) до азоту в сечі (відношення Д/А). У тварин, що голодують, глікоген печінки вичерпується, а гліцерин перетворюється в глюкозу в дуже обмеженій кількості, так що єдиним важливим джерелом глюкози плазми є білок (див. Розділ 17). Можна обчислити, що кількість вуглецю в білку, яка відповідає 1 г азоту сечовини, достатня для утворення 8,3 г глюкози. Отже, відношення Д/А ≈ 3, яке простежується під час діабету, свідчить про перетворення в глюкозу близько 33% вуглецю метаболізованого білка.
Підвищення глюконеогенезу зумовлене багатьма причинами. Глюкагон стимулює Глюконеогенез, а гіперглюкагонемія, зазвичай, виникає у разі діабету. Надниркові глюкокортикоїди також призводять до збільшення глюконеогенезу, якщо їхній рівень підвищується в серйозно хворих діабетиків. Простежується збільшене постачання амінокислот для глюконеогенезу, оскільки без інсуліну в м’язах синтезується менше білків, а отже, рівень амінокислот у крові збільшується. Аланін особливо легко перетворюється в глюкозу. Окрім цього, посилюється активність ензимів, що каталізують перетворення пірувату та інших двовуглецевих обмінних фрагментів у глюкозу. Це, зокрема, стосується фосфоенолпіруваткарбоксикінази, яка полегшує перетворення оксалоацетату у фосфоенолпіруват (див. Розділ 17), а також фруктозо-1,6-дифосфату, що каталізує перетворення фруктозодифосфату у фруктозо-6-фосфат і глюкозо-6-фосфат, який контролює надходження глюкози з печінки у кров. Збільшення ацетил-КоА стимулює піруваткарбоксилазну активність, а нестача інсуліну веде до збільшеного утворення ацетил-КоА, оскільки ліпогенез сповільнюється. Піруваткарбоксилаза каталізує перетворення пірувату в оксалоацетат (див. рис. 17-9).
Наслідок впливу прискореного перетворення білка в СO2, Н2O і глюкозу разом зі зменшеним білковим синтезом у разі діабету зумовлює негативний азотний баланс, білкову недостатність та виснаження. Білкове виснаження з будь-якої причини пов’язане з низькою резистентністю до інфекцій.
Метаболізм жирів у разі діабету
Головними аномаліями метаболізму жирів у випадку діабету є прискорення катаболізму ліпідів зі збільшеним утворенням кетонових тіл та зменшення синтезу жирних кислот і тригліцеридів. Прояви цих розладів метаболізму жирів настільки помітні, що діабет називають більше хворобою метаболізму жирів, ніж вуглеводів.
За нормальних умов відбувається спалення 50% спожитої глюкози до СO2 та Н2O; 5% її перетворюється в глікоген; а 30-40% перетворюється в жир у жирових відкладах. У разі діабету менш ніж 5% перетворюється в жир, незважаючи на те, що кількість, спалена до СO2 та Н2O, також зменшена, а кількість, що перетворюється в глікоген, не збільшується. Отже, глюкоза накопичується в крові і переходить у сечу.
Роль ліпопротеїнліпази та гормоночутливої ліпази в регулюванні метаболізму жирів розглянуто в Розділі 17. У разі діабету зменшується перетворення глюкози в Жирні кислоти за умов депонування внаслідок внутрішньоклітинного дефіциту глюкози. Інсулін інгібує гормоночутливу ліпазу в жировій тканині, і без цього гормону рівень вільних жирних кислот (ВЖК) у плазмі підвищується більше ніж удвічі. Збільшення глюкагону також сприяє мобілізації ВЖК. Отже, рівень ВЖК змінюється одночасно з рівнем глюкози в плазмі і є дещо ліпшим показником складності діабетичного стану. В печінці та інших тканинах жирні кислоти катаболізують до ацетил-КоА. Деяка частина ацетил-КоА згорає разом з амінокислотними залишками з утворенням СO2 і Н2O в циклі лимонної кислоти. Однак надходження перевищують здатність тканин катаболізувати ацетил-КоА.
Процеси, що відбуваються в печінці у разі діабету, показані на рис. 19-10. Крім уже згаданого збільшення глюконеогенезу та значного викиду глюкози в кров, також відчутно погіршується перетворення ацетил-КоА в малоніл-КоА, а з нього - у жирні кислоти. Воно виникає внаслідок нестачі ацетил-КоА карбоксилази, ензиму, що каталізує перетворення. Надлишок ацетил-КоА перетворюється в кетонові тіла (див. нижче).
У випадку нелікованого діабету збільшується концентрація тригліцеридів, хіломікронів та ВЖК у плазмі; такий стан називають ліпемією (гіперліпемією). Збільшення вмісту цих складових є наслідком зменшеного переміщення тригліцеридів у жирові відклади. Зменшення активності ліпопротеїнліпаз робить внесок у зменшене видалення тригліцеридів.
Кетонемія
Якщо в організмі є надлишок ацетил-КоА, то деяка його частина перетворюється в ацетоацетил-КоА, а потім, у печінці, в ацетоацетат. Ацетоацетат та його похідні - ацетон і ß-гідроксибутират - надходять у кров у значних кількостях (див. Розділ 17). Ці кетонові тіла, що циркулюють, є важливим джерелом енергії під час голодування. Вважають, що в нормальних собак під час голодування половина метаболізму відбувається внаслідок метаболізму кетонових тіл. У діабетиків рівень утилізації кетонових тіл також суттєвий. З’ясовано, що у хворих на діабет максимальний рівень, за якого жир може катаболізуватись без помітної кетонемії, становить 2,5 г/кг маси тіла за добу. У разі нелікованого діабету продукування набагато вище від зазначеного, і кетонові тіла накопичуються в кров’яному руслі. Є деякі докази того, що в разі важкого діабету рівень утилізації кетонових тіл може знижуватися, погіршуючи кетонемію, інсулін же збільшує використання кетонових тіл м’язами.
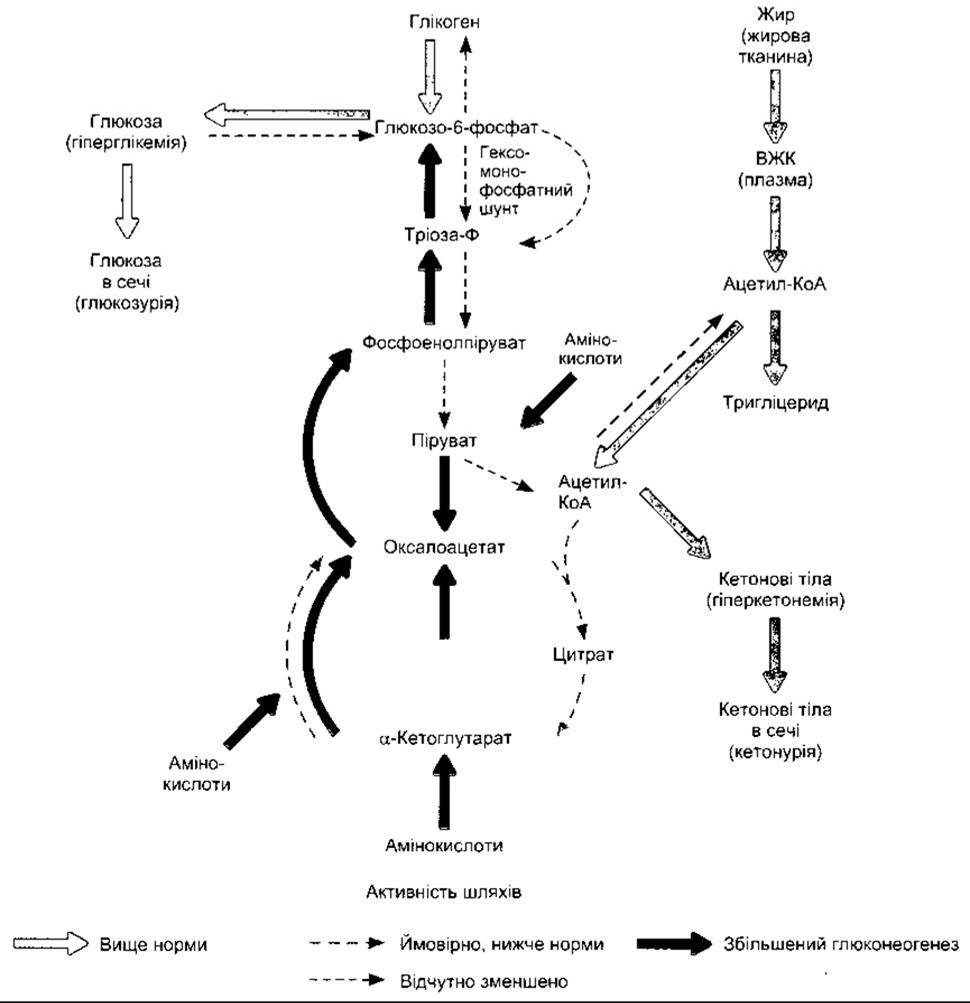
Рис. 19-10. Метаболічні аномалії в печінці у випадку нелікованого діабету (відтворено за дозволом з Murray RK et al: Harper's Biochemistry, 25th ed. McGraw-Hill, 2000).
Ацидоз
Більшість йонів водню, вивільнених з ацетоацетату та ß-гідроксибутирату, буферизують, та все ж сильний метаболічний ацидоз розвивається. Зниження pH крові стимулює дихальний центр, що веде до появи швидкого глибокого Дихання, описаного Куссмаулем як повітряний голод і названим на його честь диханням Куссмауля. У цьому випадку сеча стає кислою. Якщо здатність нирок заміщати катіони плазми, що супроводжують органічні аніони, на Н+ і NH4+ перевищена, то Na+ і К+ виходять із сечею. Втрата води та електролітів веде до дегідратації, гіповолемії та зниження кров’яного тиску. Нарешті, ацидоз та дегідратація пригнічують свідомість до стану коми. Незважаючи на те, що тепер інфекції, які ускладнюють хворобу, вдається контролювати за допомогою антибіотиків, ацидоз є найпоширенішою причиною ранньої смерті в разі цукрового діабету у клініці.
У разі вираженого ацидозу загальна кількість натрію в організмі значно зменшується, а якщо втрати натрію перевищують втрати води, то рівень Na+ в плазмі також стає низьким. Загальний рівень калію в організмі теж невисокий, однак його плазматичний рівень зазвичай нормальний частково внаслідок зменшення об’єму ПКР, а частково тому, що К+ рухається з клітин до ПКР, коли позаклітинна концентрація Н+ висока. Іншим фактором, що підтримує рівень К+ у плазмі, є відсутність інсуліноіндукованого надходження К+ у клітину.
Ступінь, з яким кетоацидоз ускладнює експериментальний діабет, змінюється в різних видів. Розмір запасів жиру в організмі також є фактором, що визначає відповідь на діабет. До виділення інсуліну Бантінгом і Бестом 1921 р. головним методом Лікування діабету в людей було голодування (режим Харчування Аллена). Це зменшувало не лише рівень глюкози в плазмі, а й депонування жиру до тої міри, коли вже було мало жиру для мобілізації.
Кома
Кома в разі діабету може виникати внаслідок ацидозу та дегідратації. Проте глюкоза в плазмі може збільшитись до такого рівня, що не залежить від pH плазми, і гіперосмолярність плазми призведе до втрати свідомості (гіперосмолярна кома). Накопичення лактату в крові (лактоцидоз) також ускладнює діабетичний кетоацидоз, якщо тканини стають гіпоксичними (див. Розділ 33), і може призвести до коми. Набряк мозку простежується в значної кількості пацієнтів із діабетичним ацидозом, він також може призвести до коми. Причина набряку мозку остаточно не з’ясована, однак це серйозне ускладнення з поганим прогнозом.
Метаболізм холестеролу
У разі діабету характерне підвищення рівня холестеролу в плазмі, що прискорює розвиток судинної хвороби атеросклерозу, яка є головним довготривалим ускладненням діабету в людини. Рівень холестеролу підвищується внаслідок збільшення концентрації ЛПДНЩ та ЛПНЩ у плазмі (див. Розділ 17), що може бути наслідком збільшеного утворення ЛПДНЩ у печінці чи зменшеного видалення ЛПДНЩ та ЛПНЩ із кров’яного русла.
Складність метаболічних порушень у разі діабету потребує підсумування описаних вище положень. Однією з ключових ознак дефіциту інсуліну (рис. 19-11) є зменшене надходження глюкози до багатьох тканин (зменшена периферійна утилізація). Простежується також збільшення чистого вивільнення глюкози з печінки (збільшене утворення) частково внаслідок надлишку глюкагону. Як наслідок, гіперглікемія веде до глюкозурії та дегідратаційного осмотичного діурезу. Дегідратація спричинює полідипсію. На тлі внутрішньоклітинної нестачі глюкози стимульований апетит; глюкоза утворюється з білків (глюконеогенез), а енергетичні потреби підтримує Метаболізм білків та жирів. Результатом цього є втрата маси, слабкість, дефіцит білків та виснаження. Катаболізм жирів посилюється, і в організмі виникає надлишок тригліцеридів і ВЖК. Синтез жирів пригнічений, а перевантажені катаболічні шляхи не можуть дати собі раду з надлишком утвореного ацетил-КоА. У печінці ацетил-КоА перетворюється в кетонові тіла. Кетонові тіла, головно, є органічними кислотами, які накопичуються в крові (кетонемія), оскільки швидкість їхнього утворення перевищує здатність організму до їхньої утилізації. З накопиченням кетонових тіл розвивається метаболічний ацидоз. Зменшення кількості Na+ і К+ супроводжує дегідратація, оскільки катіони плазми екскретуються з органічними аніонами, які не з’єднуються з Н+ і NH4+, що їх секретують нирки. Зрештою, ацидоз, гіповолемія, гіпотензія виснаженої тварини чи пацієнта зумовлюють стан коми внаслідок токсичних ефектів ацидозу, дегідратації та гіперосмолярності на нервову систему і, якщо їм вчасно не надано медичну допомогу, - смерть.

Рис. 19-11. Ефекти нестачі інсуліну (з дозволу RJ Havel).
Усі ці порушення коректують уведенням інсуліну. Хоча негайне лікування ацидозу також передбачає парентеральне олужнення для боротьби з ацидозом та наводнення для поповнення запасів Na+ і К+ організму, однак лише інсулін виліковує головні дефекти, уможливлюючи повернення до нормального стану.
Последнее обновление: 05/02/2024
Редакционная и учебная адаптация: Данный материал сведен на основе первоисточника/оригинального текста. Команда проекта осуществила редакционную обзорную обработку, исправление технических неточностей, структурирование разделов и адаптацию содержания к учебному формату.
Что было обработано:
- устранение форматных дефектов (OCR-ошибки, разрывы структуры, дефектные символы);
- редакционное упорядочивание содержания;
- унификация терминов в соответствии с академическими источниками;
- проверка соответствия фактических утверждений текста первоисточнику.
Все упоминания об авторе, годе издания и происхождении первичного текста сохранены в соответствии с источником.